Sun Rui, Sode Olaseni, Dama James F, Voth Gregory A
Department of Chemistry, James Franck Institute, and Institute for Biophysical Dynamics, The University of Chicago , 5735 South Ellis Avenue, Chicago, Illinois 60637, United States.
J Chem Theory Comput. 2017 May 9;13(5):2332-2341. doi: 10.1021/acs.jctc.7b00077. Epub 2017 Apr 4.
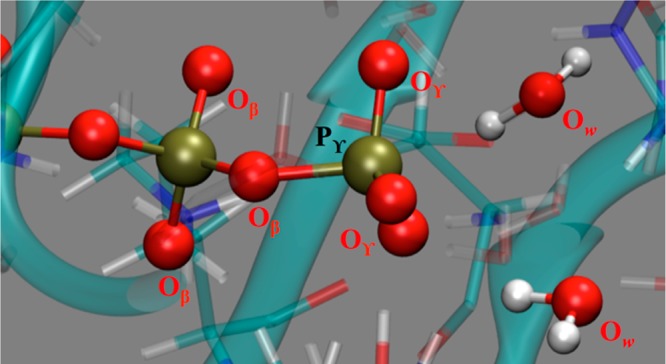
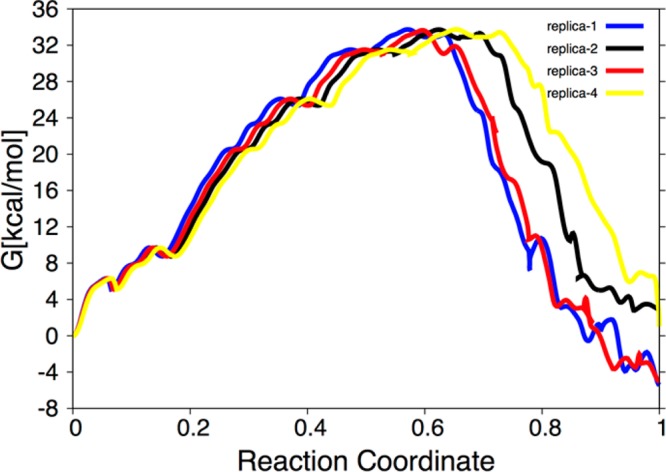
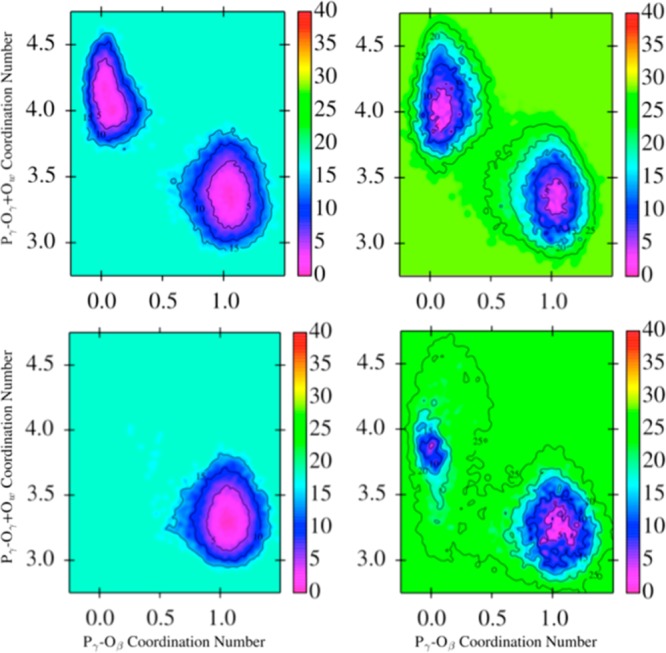
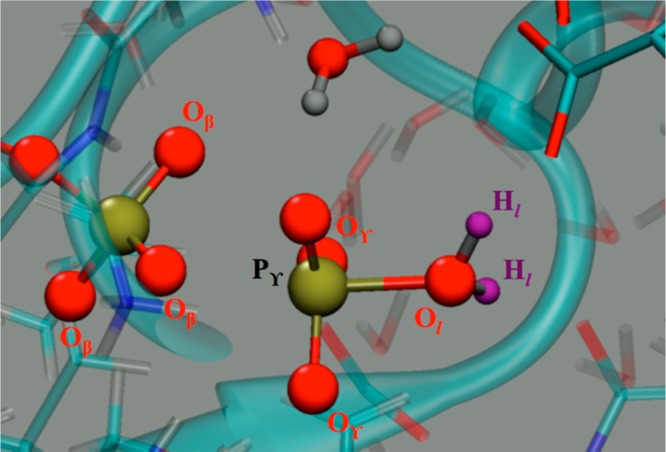
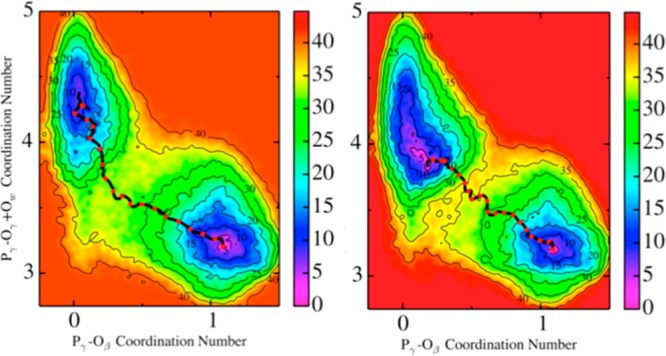
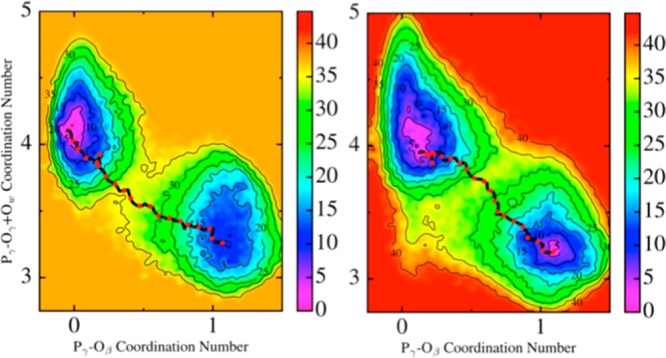
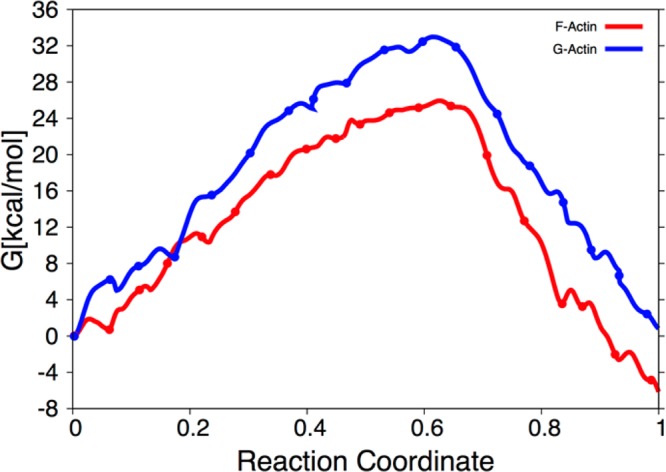
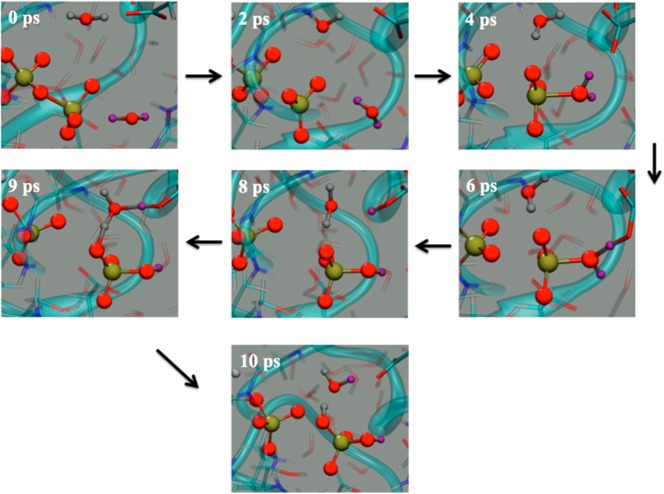
The protein mediated hydrolysis of nucleoside triphosphates such as ATP or GTP is one of the most important and challenging biochemical reactions in nature. The chemical environment (water structure, catalytic metal, and amino acid residues) adjacent to the hydrolysis site contains hundreds of atoms, usually greatly limiting the amount of the free energy sampling that one can achieve from computationally demanding electronic structure calculations such as QM/MM simulations. Therefore, the combination of QM/MM molecular dynamics with the recently developed transition-tempered metadynamics (TTMetaD), an enhanced sampling method that can provide a high-quality free energy estimate at an early stage in a simulation, is an ideal approach to address the biomolecular nucleoside triphosphate hydrolysis problem. In this work the ATP hydrolysis process in monomeric and filamentous actin is studied as an example application of the combined methodology. The performance of TTMetaD in these demanding QM/MM simulations is compared with that of the more conventional well-tempered metadynamics (WTMetaD). Our results show that TTMetaD exhibits much better exploration of the hydrolysis reaction free energy surface in two key collective variables (CVs) during the early stages of the QM/MM simulation than does WTMetaD. The TTMetaD simulations also reveal that a key third degree of freedom, the O-H bond-breaking and proton transfer from the lytic water, must be biased for TTMetaD to converge fully. To perturb the NTP hydrolysis dynamics to the least extent and to properly focus the MetaD free energy sampling, we also adopt here the recently developed metabasin metadynamics (MBMetaD) to construct a self-limiting bias potential that only applies to the lytic water after its nucleophilic attack of the phosphate of ATP. With these new, state-of-the-art enhanced sampling metadynamics techniques, we present an effective and accurate computational strategy for combining QM/MM molecular dynamics simulation with free energy sampling methodology, including a means to analyze the convergence of the calculations through robust numerical criteria.
蛋白质介导的三磷酸核苷(如ATP或GTP)水解是自然界中最重要且最具挑战性的生化反应之一。水解位点附近的化学环境(水结构、催化金属和氨基酸残基)包含数百个原子,这通常极大地限制了通过诸如QM/MM模拟等计算量很大的电子结构计算所能实现的自由能采样量。因此,将QM/MM分子动力学与最近开发的过渡温度元动力学(TTMetaD)相结合是解决生物分子三磷酸核苷水解问题的理想方法,TTMetaD是一种增强采样方法,能够在模拟早期提供高质量的自由能估计。在这项工作中,以单体和丝状肌动蛋白中的ATP水解过程作为该组合方法的示例应用进行研究。将TTMetaD在这些高要求的QM/MM模拟中的性能与更传统的加权温度元动力学(WTMetaD)的性能进行了比较。我们的结果表明,在QM/MM模拟的早期阶段,TTMetaD在两个关键的集体变量(CVs)中对水解反应自由能表面的探索比WTMetaD要好得多。TTMetaD模拟还表明,对于TTMetaD要完全收敛,一个关键的第三自由度,即来自裂解水的O-H键断裂和质子转移,必须进行偏差设定。为了在最小程度上扰动NTP水解动力学并恰当地聚焦元动力学自由能采样,我们在此还采用了最近开发的元盆地元动力学(MBMetaD)来构建一个自限偏差势,该偏差势仅在裂解水对ATP的磷酸基团进行亲核攻击后才应用于裂解水。借助这些新的、最先进的增强采样元动力学技术,我们提出了一种将QM/MM分子动力学模拟与自由能采样方法相结合的有效且准确的计算策略,包括一种通过稳健的数值标准来分析计算收敛性的方法。