Paulsen Janet L, Leidner Florian, Ragland Debra A, Kurt Yilmaz Nese, Schiffer Celia A
Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School , Worcester, Massachusetts 01605, United States.
J Chem Theory Comput. 2017 May 9;13(5):2300-2309. doi: 10.1021/acs.jctc.6b01262. Epub 2017 Apr 11.
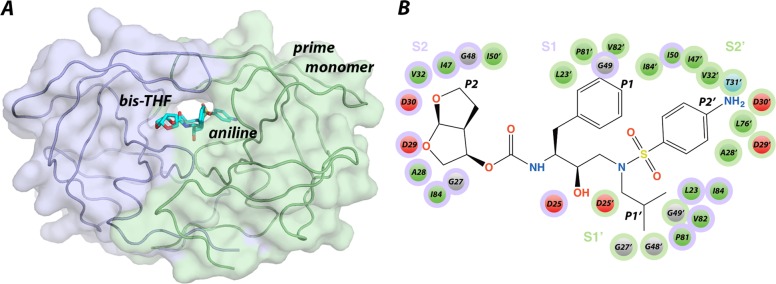
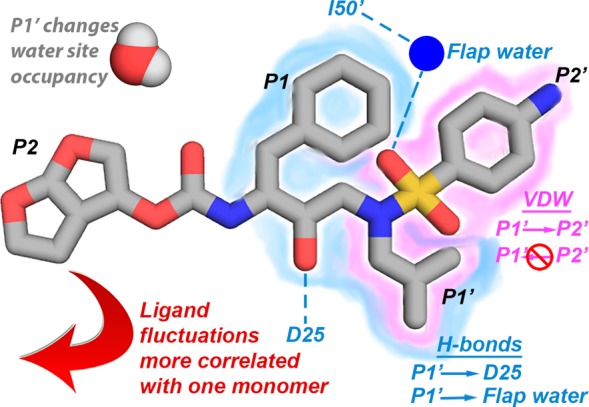
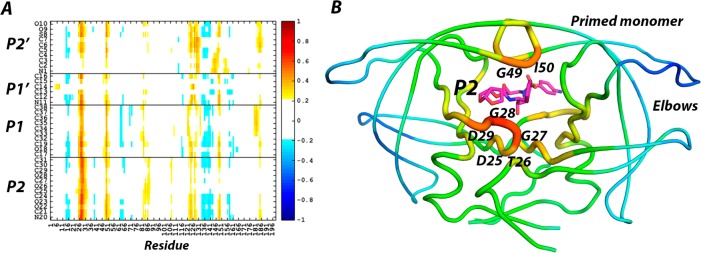
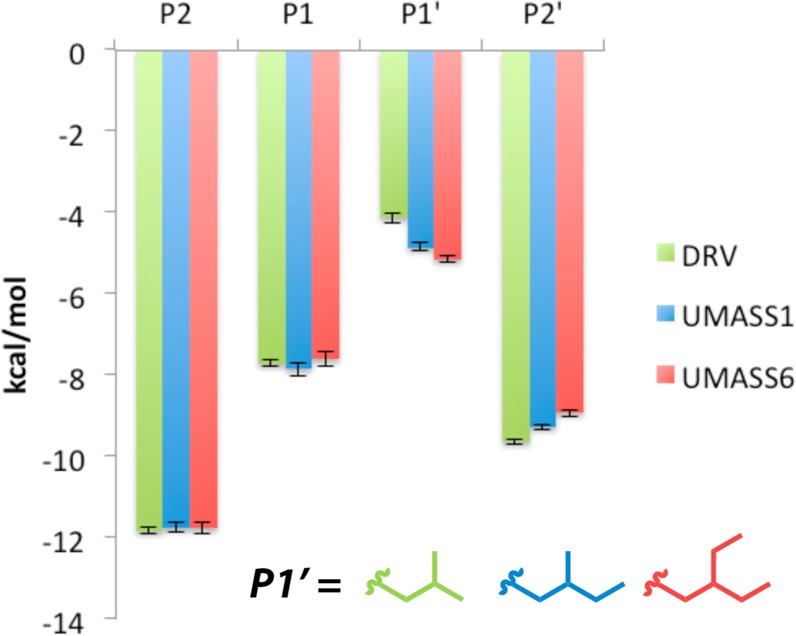
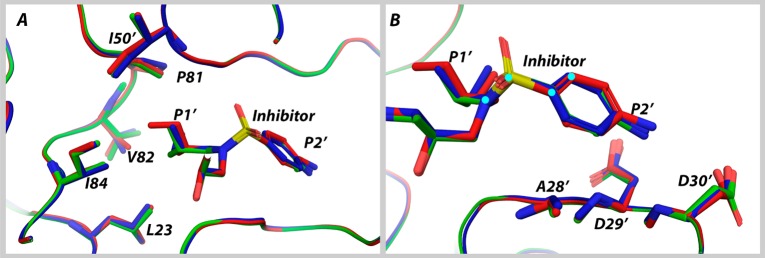
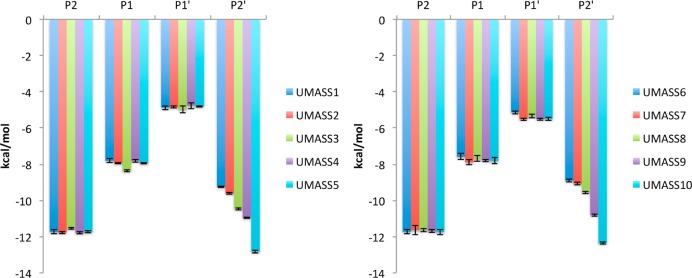
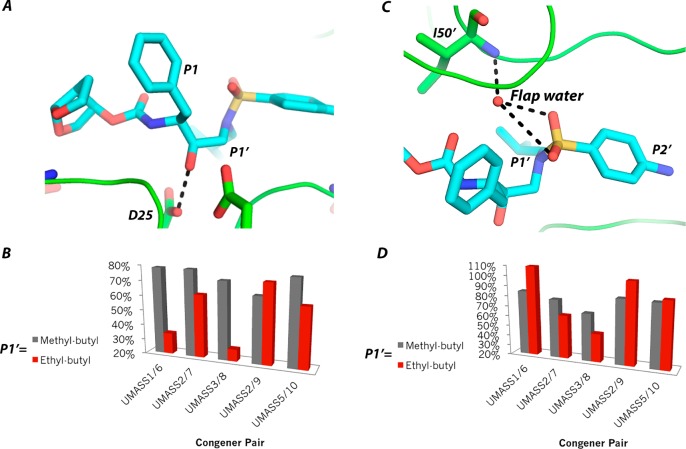
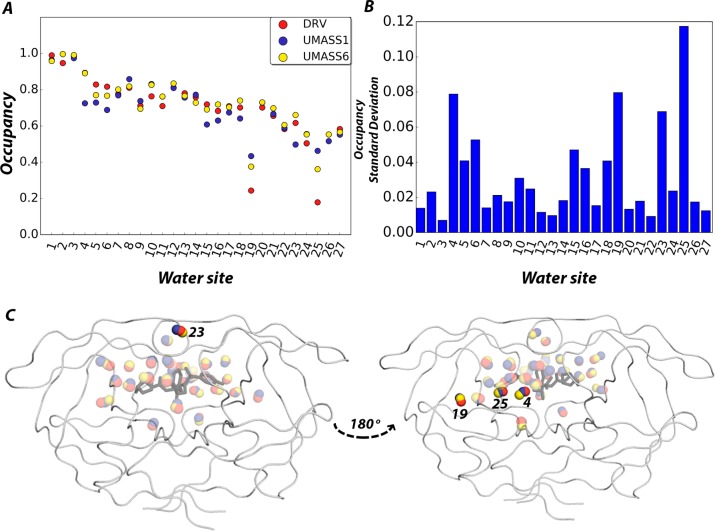
Molecular recognition is a highly interdependent process. Subsite couplings within the active site of proteases are most often revealed through conditional amino acid preferences in substrate recognition. However, the potential effect of these couplings on inhibition and thus inhibitor design is largely unexplored. The present study examines the interdependency of subsites in HIV-1 protease using a focused library of protease inhibitors, to aid in future inhibitor design. Previously a series of darunavir (DRV) analogs was designed to systematically probe the S1' and S2' subsites. Co-crystal structures of these analogs with HIV-1 protease provide the ideal opportunity to probe subsite interdependency. All-atom molecular dynamics simulations starting from these structures were performed and systematically analyzed in terms of atomic fluctuations, intermolecular interactions, and water structure. These analyses reveal that the S1' subsite highly influences other subsites: the extension of the hydrophobic P1' moiety results in 1) reduced van der Waals contacts in the P2' subsite, 2) more variability in the hydrogen bond frequencies with catalytic residues and the flap water, and 3) changes in the occupancy of conserved water sites both proximal and distal to the active site. In addition, one of the monomers in this homodimeric enzyme has atomic fluctuations more highly correlated with DRV than the other monomer. These relationships intricately link the HIV-1 protease subsites and are critical to understanding molecular recognition and inhibitor binding. More broadly, the interdependency of subsite recognition within an active site requires consideration in the selection of chemical moieties in drug design; this strategy is in contrast to what is traditionally done with independent optimization of chemical moieties of an inhibitor.
分子识别是一个高度相互依赖的过程。蛋白酶活性位点内的亚位点偶联最常通过底物识别中的条件性氨基酸偏好来揭示。然而,这些偶联对抑制作用以及抑制剂设计的潜在影响在很大程度上尚未得到探索。本研究使用聚焦的蛋白酶抑制剂文库研究了HIV-1蛋白酶中亚位点的相互依赖性,以辅助未来的抑制剂设计。此前设计了一系列达芦那韦(DRV)类似物来系统地探测S1'和S2'亚位点。这些类似物与HIV-1蛋白酶的共晶体结构为探测亚位点相互依赖性提供了理想的机会。从这些结构出发进行了全原子分子动力学模拟,并根据原子波动、分子间相互作用和水结构进行了系统分析。这些分析表明,S1'亚位点对其他亚位点有高度影响:疏水P1'部分的延伸导致1)P2'亚位点中范德华接触减少,2)与催化残基和瓣状水的氢键频率有更大的变异性,以及3)活性位点近端和远端保守水位点占有率的变化。此外,这种同二聚体酶中的一个单体的原子波动与DRV的相关性比另一个单体更高。这些关系错综复杂地连接了HIV-1蛋白酶的亚位点,对于理解分子识别和抑制剂结合至关重要。更广泛地说,在药物设计中选择化学基团时需要考虑活性位点内亚位点识别的相互依赖性;这种策略与传统上对抑制剂化学基团进行独立优化的做法形成对比。