Argyropoulos Christos, Etheridge Alton, Sakhanenko Nikita, Galas David
Department of Internal Medicine, University of New Mexico School of Medicine, Albuquerque, NM 87106, USA.
Pacific Northwest Research Institute, Seattle, WA 98122, USA.
Nucleic Acids Res. 2017 Jun 20;45(11):e104. doi: 10.1093/nar/gkx199.
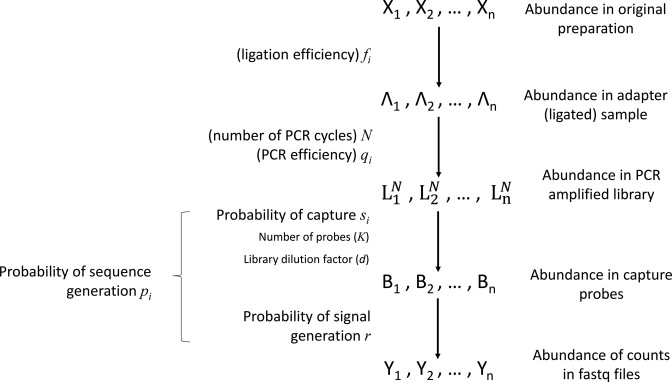
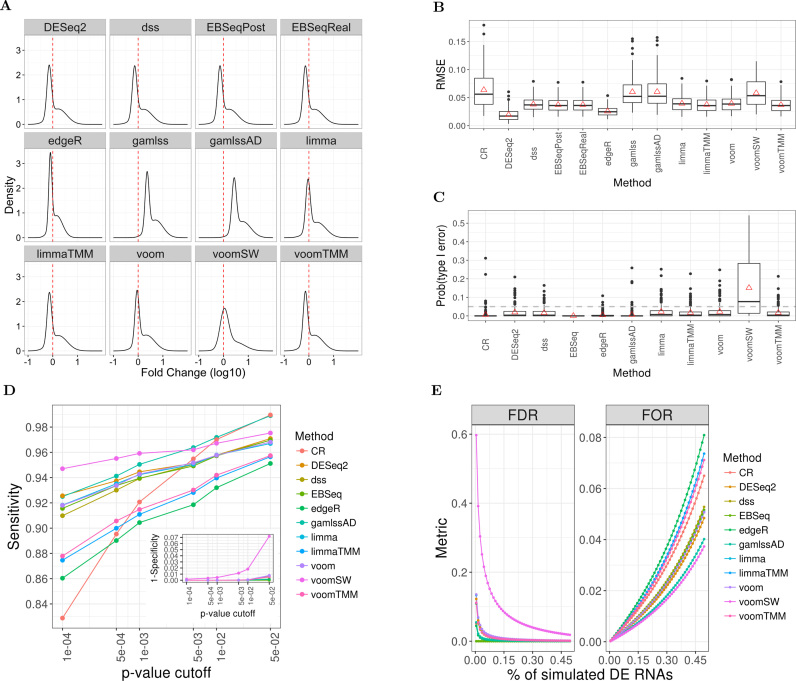
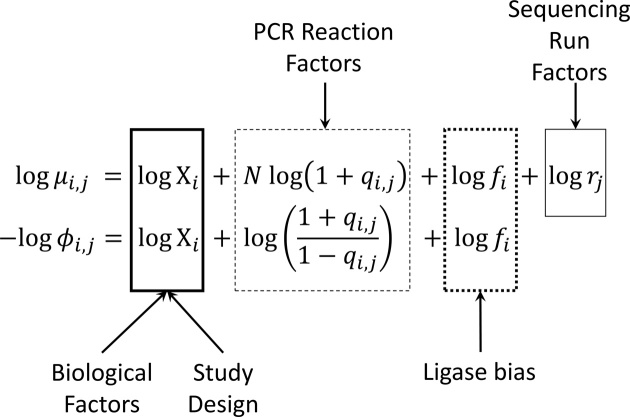
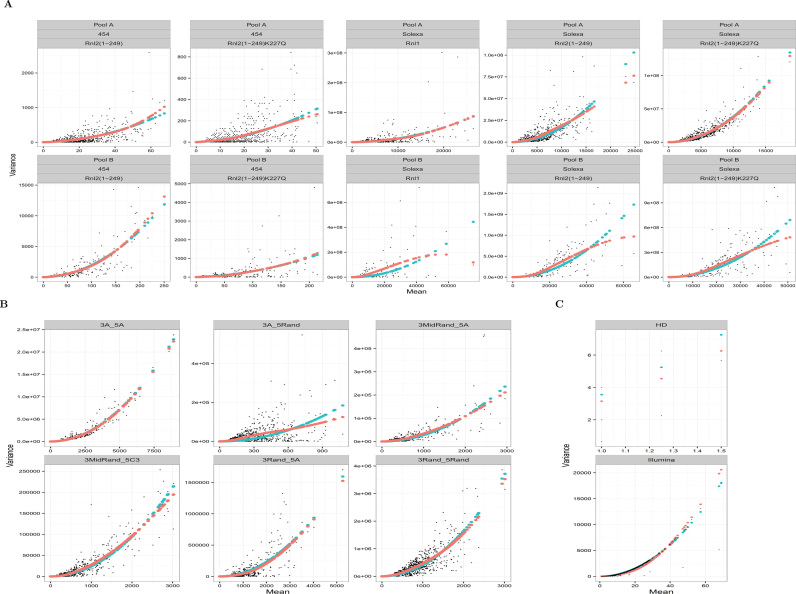
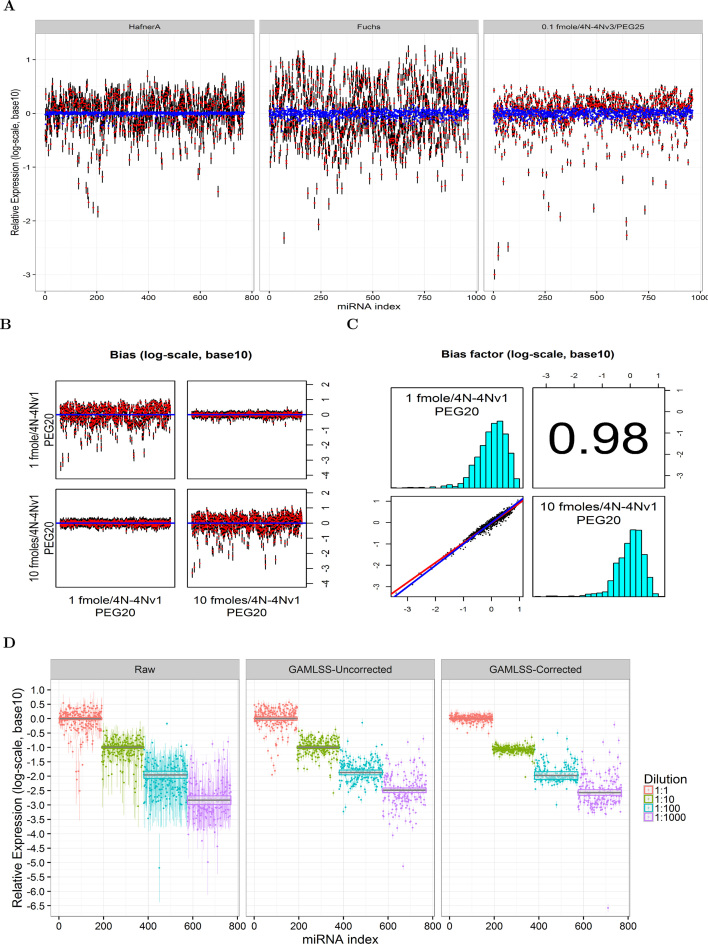
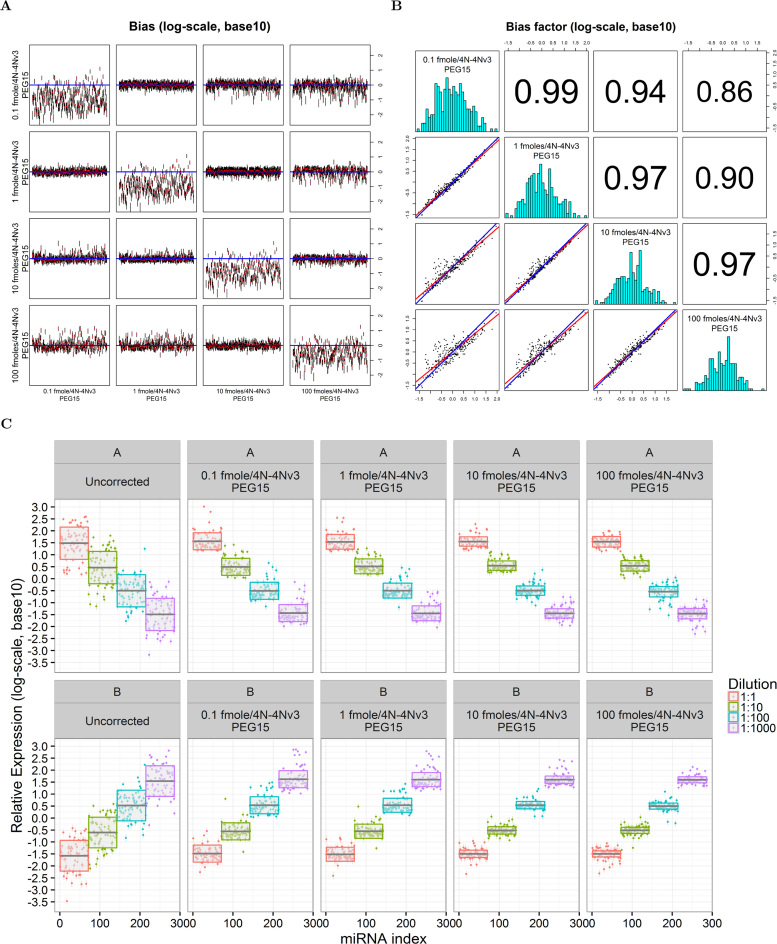
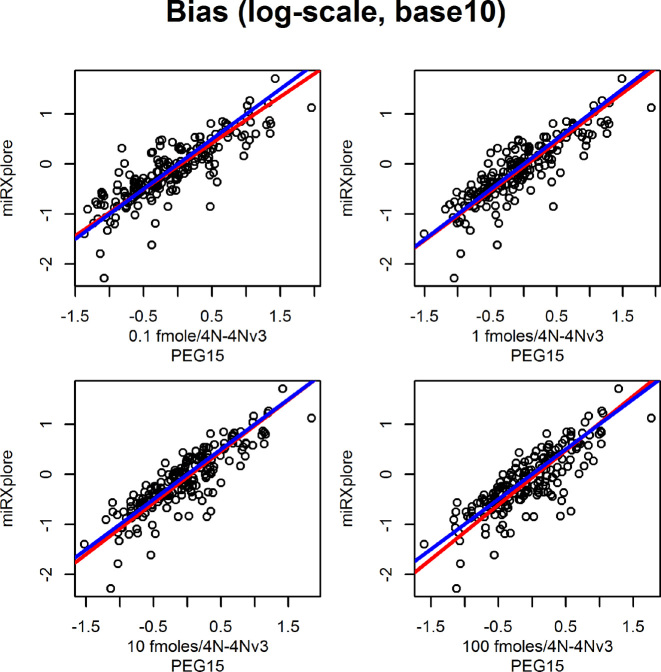
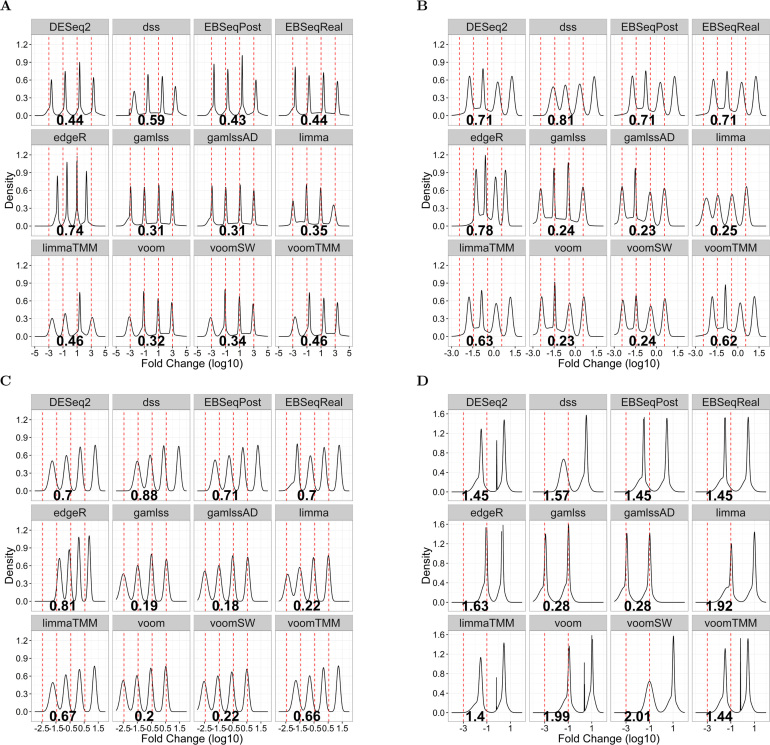
The use of RNA-seq as the preferred method for the discovery and validation of small RNA biomarkers has been hindered by high quantitative variability and biased sequence counts. In this paper we develop a statistical model for sequence counts that accounts for ligase bias and stochastic variation in sequence counts. This model implies a linear quadratic relation between the mean and variance of sequence counts. Using a large number of sequencing datasets, we demonstrate how one can use the generalized additive models for location, scale and shape (GAMLSS) distributional regression framework to calculate and apply empirical correction factors for ligase bias. Bias correction could remove more than 40% of the bias for miRNAs. Empirical bias correction factors appear to be nearly constant over at least one and up to four orders of magnitude of total RNA input and independent of sample composition. Using synthetic mixes of known composition, we show that the GAMLSS approach can analyze differential expression with greater accuracy, higher sensitivity and specificity than six existing algorithms (DESeq2, edgeR, EBSeq, limma, DSS, voom) for the analysis of small RNA-seq data.
RNA测序作为发现和验证小RNA生物标志物的首选方法,一直受到高定量变异性和序列计数偏差的阻碍。在本文中,我们开发了一种针对序列计数的统计模型,该模型考虑了连接酶偏差和序列计数中的随机变异。该模型意味着序列计数的均值和方差之间存在线性二次关系。使用大量测序数据集,我们展示了如何使用位置、尺度和形状的广义相加模型(GAMLSS)分布回归框架来计算和应用连接酶偏差的经验校正因子。偏差校正可以消除超过40%的miRNA偏差。经验偏差校正因子在至少一个到四个数量级的总RNA输入范围内似乎几乎恒定,并且与样品组成无关。使用已知组成的合成混合物,我们表明,对于小RNA测序数据分析,GAMLSS方法比六种现有算法(DESeq2、edgeR、EBSeq、limma、DSS、voom)能够更准确、更灵敏且更具特异性地分析差异表达。