Department of Integrative Biology, Oregon State University, Corvallis, Oregon 97331
Department of Integrative Biology, Oregon State University, Corvallis, Oregon 97331.
G3 (Bethesda). 2017 Jul 5;7(7):2353-2361. doi: 10.1534/g3.117.041319.
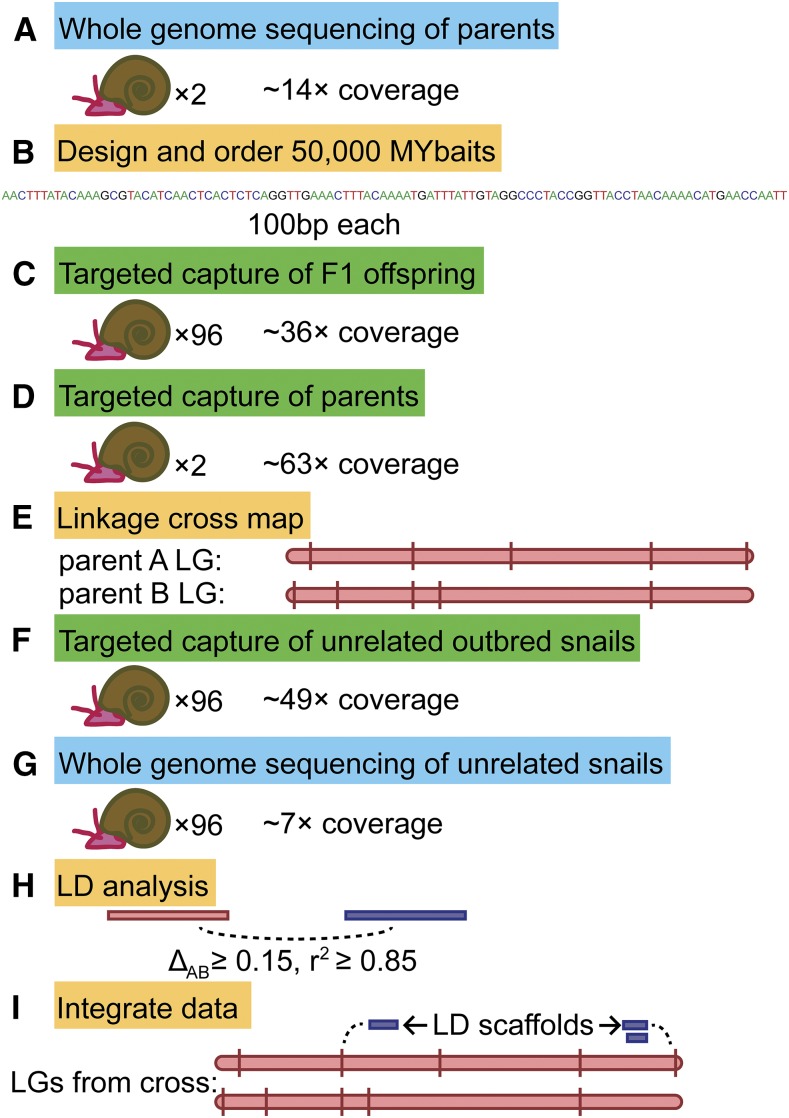
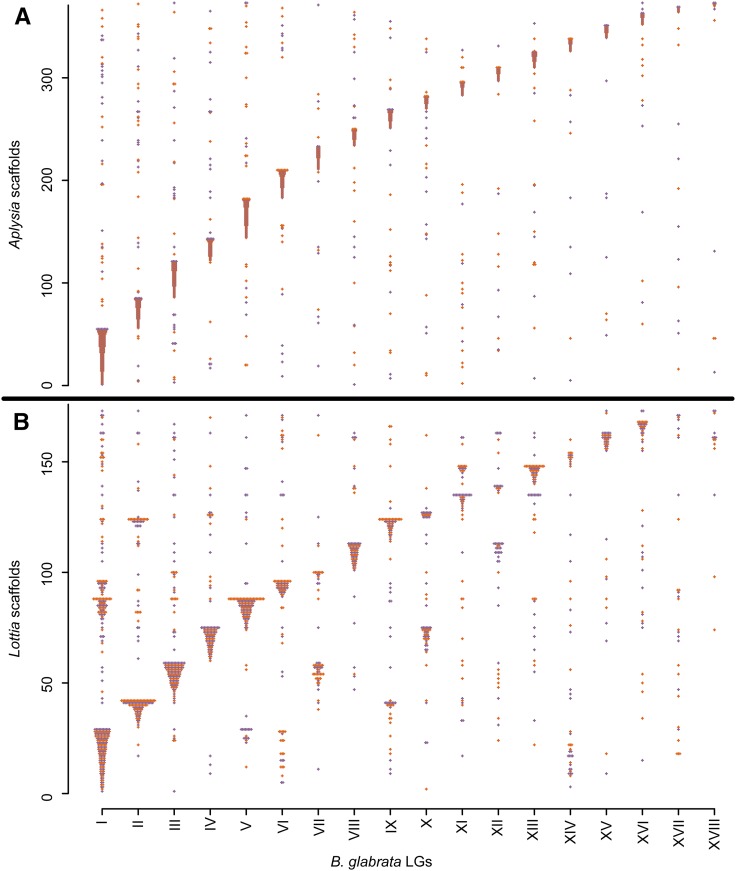
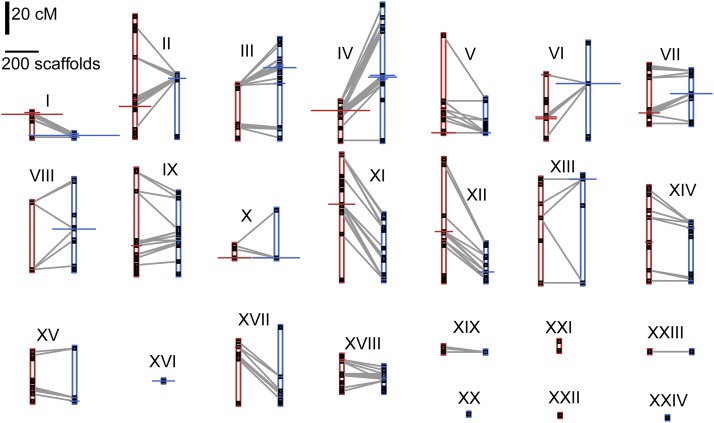
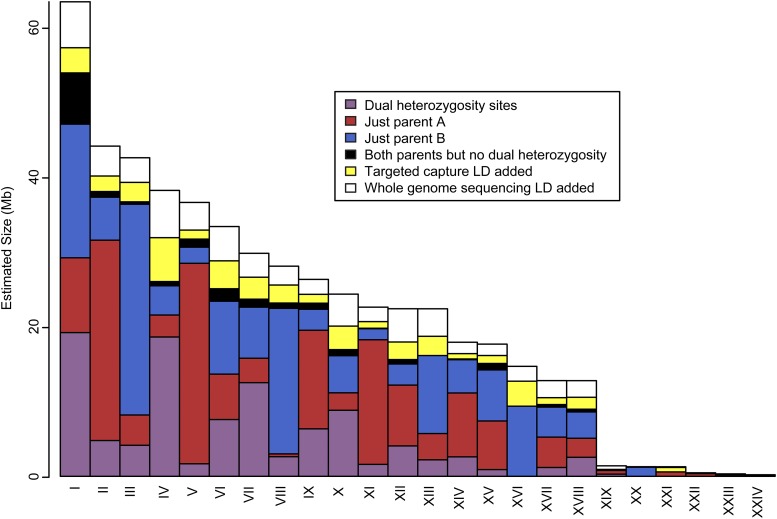
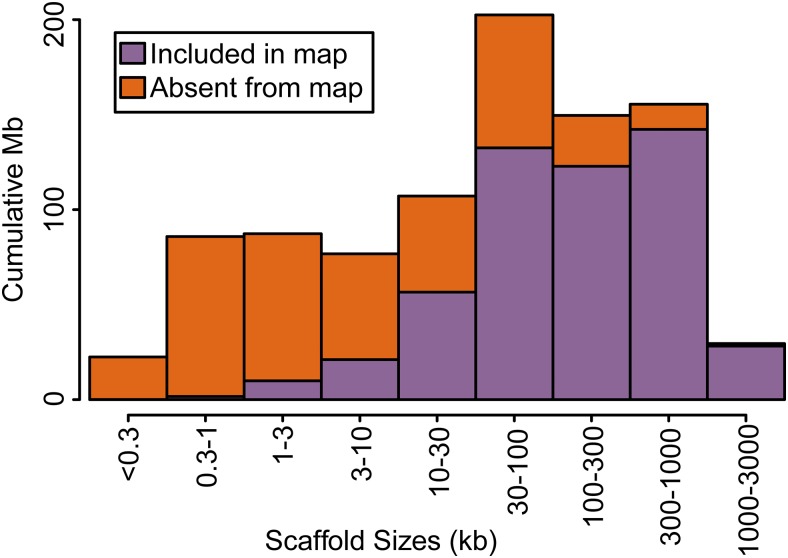
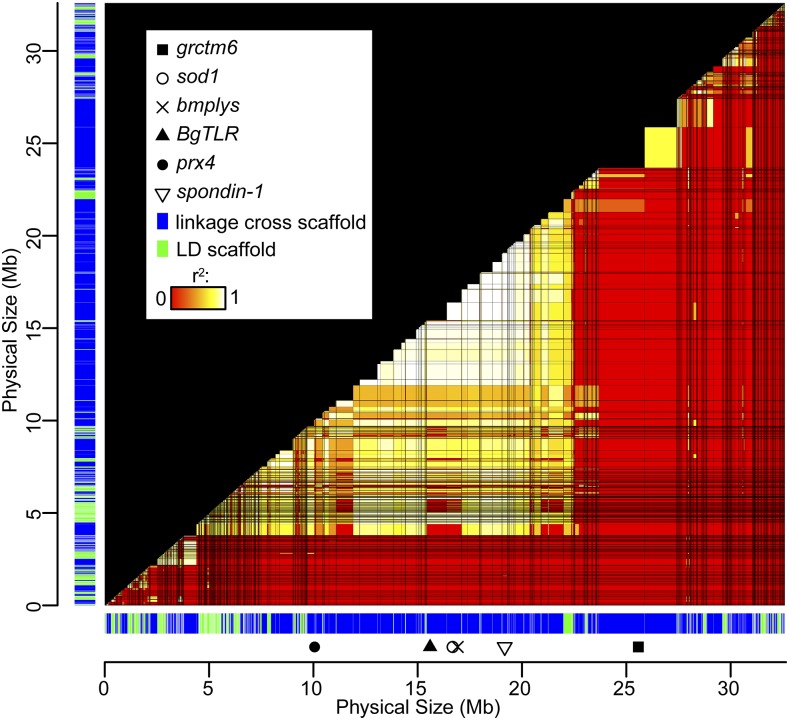
The aquatic planorbid snail is one of the most intensively-studied mollusks due to its role in the transmission of schistosomiasis. Its 916 Mb genome has recently been sequenced and annotated, but it remains poorly assembled. Here, we used targeted capture markers to map over 10,000 scaffolds in a linkage cross of 94 F1 offspring, generating 24 linkage groups (LGs). We added additional scaffolds to these LGs based on linkage disequilibrium (LD) analysis of targeted capture and whole-genome sequences of 96 unrelated snails. Our final linkage map consists of 18,613 scaffolds comprising 515 Mb, representing 56% of the genome and 75% of genic and nonrepetitive regions. There are 18 large (> 10 Mb) LGs, likely representing the expected 18 haploid chromosomes, and > 50% of the genome has been assigned to LGs of at least 17 Mb. Comparisons with other gastropod genomes reveal patterns of synteny and chromosomal rearrangements. Linkage relationships of key immune-relevant genes may help clarify snail-schistosome interactions. By focusing on linkage among genic and nonrepetitive regions, we have generated a useful resource for associating snail phenotypes with causal genes, even in the absence of a complete genome assembly. A similar approach could potentially improve numerous poorly-assembled genomes in other taxa. This map will facilitate future work on this host of a serious human parasite.
水生扁蜗牛是研究最为深入的软体动物之一,因为它在血吸虫病的传播中发挥了作用。它的 916Mb 基因组最近已经被测序和注释,但仍然组装得很差。在这里,我们使用靶向捕获标记对 94 个 F1 后代的连锁交叉中的 10000 多个支架进行了作图,生成了 24 个连锁群(LG)。我们根据 96 个无关蜗牛的靶向捕获和全基因组序列的连锁不平衡(LD)分析,将额外的支架添加到这些 LG 中。我们最终的连锁图谱由 18613 个支架组成,包含 515Mb,代表基因组的 56%和基因和非重复区域的 75%。有 18 个大的(>10Mb)LG,可能代表预期的 18 条单倍体染色体,超过 50%的基因组已被分配到至少 17Mb 的 LG 上。与其他腹足纲动物基因组的比较揭示了同线性和染色体重排的模式。关键免疫相关基因的连锁关系可能有助于阐明蜗牛-血吸虫相互作用。通过关注基因和非重复区域之间的连锁关系,我们生成了一个有用的资源,即使在没有完整基因组组装的情况下,也可以将蜗牛表型与因果基因联系起来。这种类似的方法可能会改善其他分类群中许多组装较差的基因组。这个图谱将有助于未来对这种严重的人类寄生虫宿主的研究。