Pennance Tom, Calvelo Javier, Tennessen Jacob A, Burd Ryan, Cayton Jared, Bollmann Stephanie R, Blouin Michael S, Spaan Johannie M, Hoffmann Federico G, Ogara George, Rawago Fredrick, Andiego Kennedy, Mulonga Boaz, Odhiambo Meredith, Loker Eric S, Laidemitt Martina R, Lu Lijun, Iriarte Andrés, Odiere Maurice R, Steinauer Michelle L
College of Osteopathic Medicine of the Pacific - Northwest, Western University of Health Sciences, Lebanon, OR, USA.
Laboratorio de Biología Computacional, Departamento de Desarrollo Biotecnológico, Facultad de Medicina, Instituto de Higiene, Universidad de La República, Montevideo, 11600, Uruguay.
BMC Genomics. 2024 Feb 19;25(1):192. doi: 10.1186/s12864-024-10103-w.
Control and elimination of schistosomiasis is an arduous task, with current strategies proving inadequate to break transmission. Exploration of genetic approaches to interrupt Schistosoma mansoni transmission, the causative agent for human intestinal schistosomiasis in sub-Saharan Africa and South America, has led to genomic research of the snail vector hosts of the genus Biomphalaria. Few complete genomic resources exist, with African Biomphalaria species being particularly underrepresented despite this being where the majority of S. mansoni infections occur. Here we generate and annotate the first genome assembly of Biomphalaria sudanica sensu lato, a species responsible for S. mansoni transmission in lake and marsh habitats of the African Rift Valley. Supported by whole-genome diversity data among five inbred lines, we describe orthologs of immune-relevant gene regions in the South American vector B. glabrata and present a bioinformatic pipeline to identify candidate novel pathogen recognition receptors (PRRs).
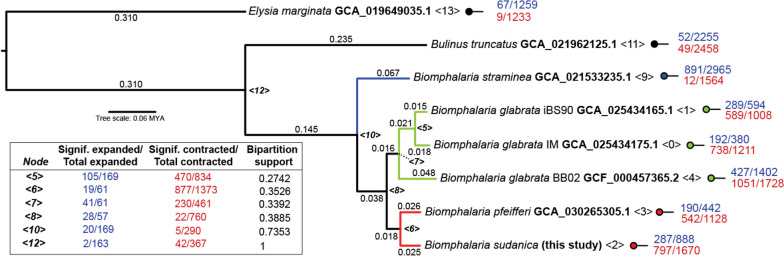
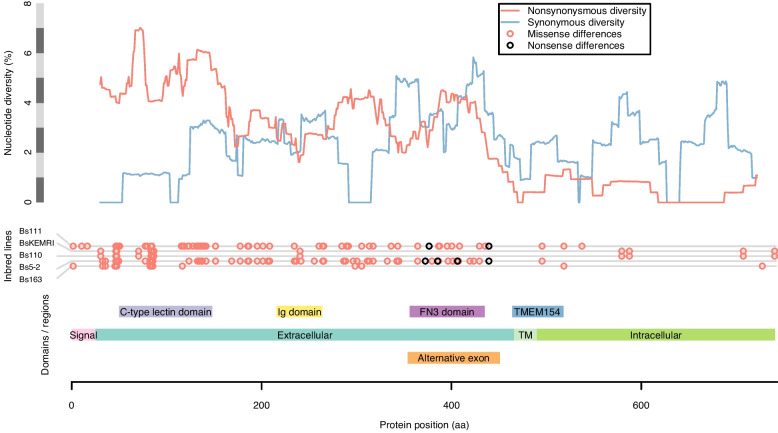
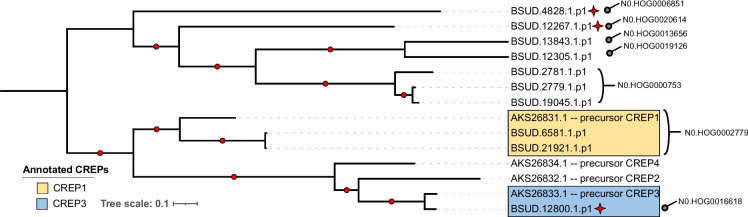
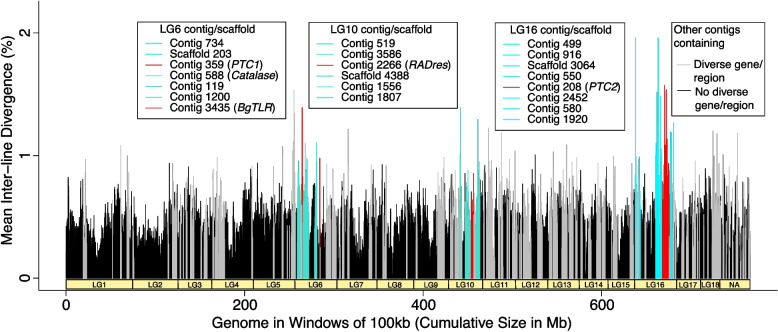
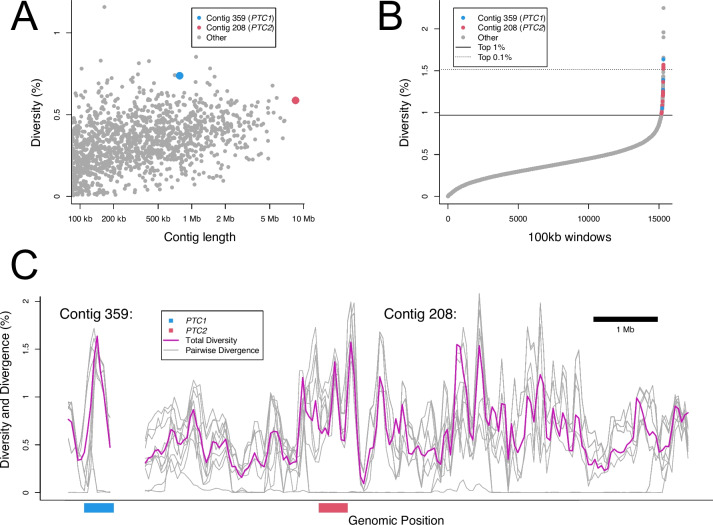
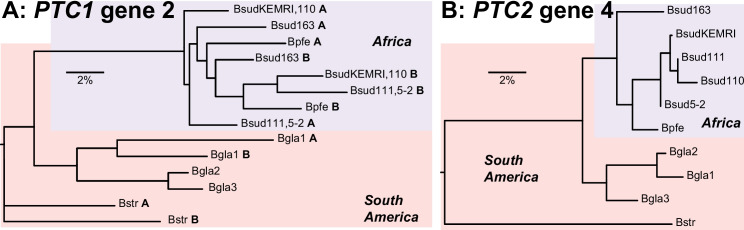
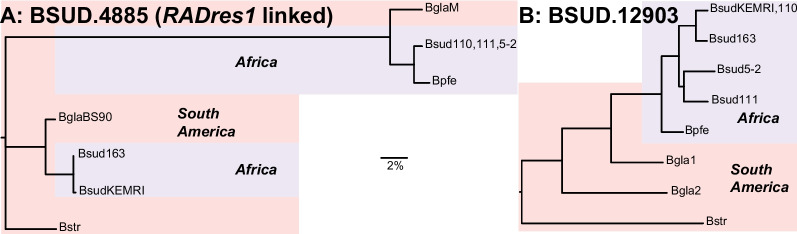
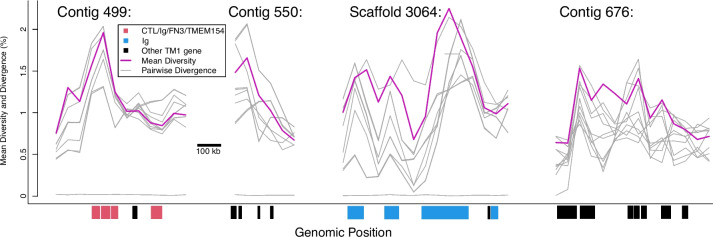
De novo genome and transcriptome assembly of inbred B. sudanica originating from the shoreline of Lake Victoria (Kisumu, Kenya) resulted in a haploid genome size of ~ 944.2 Mb (6,728 fragments, N50 = 1.067 Mb), comprising 23,598 genes (BUSCO = 93.6% complete). The B. sudanica genome contains orthologues to all described immune genes/regions tied to protection against S. mansoni in B. glabrata, including the polymorphic transmembrane clusters (PTC1 and PTC2), RADres, and other loci. The B. sudanica PTC2 candidate immune genomic region contained many PRR-like genes across a much wider genomic region than has been shown in B. glabrata, as well as a large inversion between species. High levels of intra-species nucleotide diversity were seen in PTC2, as well as in regions linked to PTC1 and RADres orthologues. Immune related and putative PRR gene families were significantly over-represented in the sub-set of B. sudanica genes determined as hyperdiverse, including high extracellular diversity in transmembrane genes, which could be under pathogen-mediated balancing selection. However, no overall expansion in immunity related genes was seen in African compared to South American lineages.
The B. sudanica genome and analyses presented here will facilitate future research in vector immune defense mechanisms against pathogens. This genomic/transcriptomic resource provides necessary data for the future development of molecular snail vector control/surveillance tools, facilitating schistosome transmission interruption mechanisms in Africa.
控制和消除血吸虫病是一项艰巨的任务,目前的策略已证明不足以阻断传播。探索通过基因方法来阻断曼氏血吸虫的传播,这种寄生虫是撒哈拉以南非洲和南美洲人类肠道血吸虫病的病原体,这引发了对双脐螺属蜗牛媒介宿主的基因组研究。完整的基因组资源很少,尽管大多数曼氏血吸虫感染发生在非洲,但非洲双脐螺物种的代表性尤其不足。在此,我们生成并注释了苏丹双脐螺广义种的首个基因组组装,该物种在非洲裂谷湖和沼泽栖息地传播曼氏血吸虫。在五个近交系全基因组多样性数据的支持下,我们描述了南美媒介光滑双脐螺中免疫相关基因区域的直系同源物,并提出了一种生物信息学流程来识别候选新型病原体识别受体(PRR)。
对源自维多利亚湖(肯尼亚基苏木)湖岸的近交苏丹双脐螺进行从头基因组和转录组组装,得到单倍体基因组大小约为944.2 Mb(6728个片段,N50 = 1.067 Mb),包含23598个基因(BUSCO = 93.6%完整)。苏丹双脐螺基因组包含与光滑双脐螺中所有已描述的针对曼氏血吸虫的免疫基因/区域的直系同源物,包括多态性跨膜簇(PTC1和PTC2)、RADres及其他位点。苏丹双脐螺PTC2候选免疫基因组区域包含许多PRR样基因,其所在基因组区域比光滑双脐螺中显示的要宽得多,并且物种间存在一个大的倒位。在PTC2以及与PTC1和RADres直系同源物相关的区域中观察到高水平的种内核苷酸多样性。在被确定为高度多样化的苏丹双脐螺基因子集中,免疫相关和假定的PRR基因家族显著富集,包括跨膜基因中的高细胞外多样性,这可能受到病原体介导的平衡选择。然而,与南美谱系相比,非洲谱系中未观察到免疫相关基因的整体扩增。
本文呈现的苏丹双脐螺基因组及分析将促进未来对媒介针对病原体的免疫防御机制的研究。这种基因组/转录组资源为未来开发分子蜗牛媒介控制/监测工具提供了必要数据,有助于非洲的血吸虫传播阻断机制研究。