Department of Plant Biology, University of California at Davis, California 95616.
Department of Botany, University of Wyoming, Laramie, Wyoming 82072.
G3 (Bethesda). 2017 Jul 5;7(7):2259-2270. doi: 10.1534/g3.117.043000.
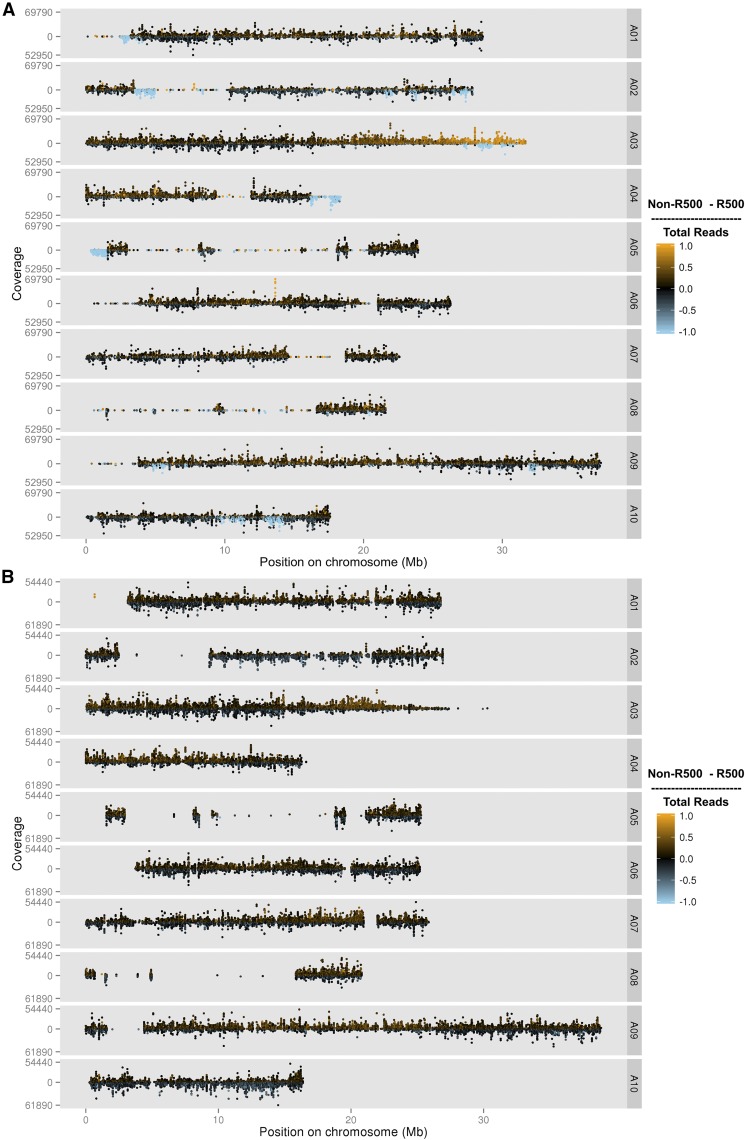
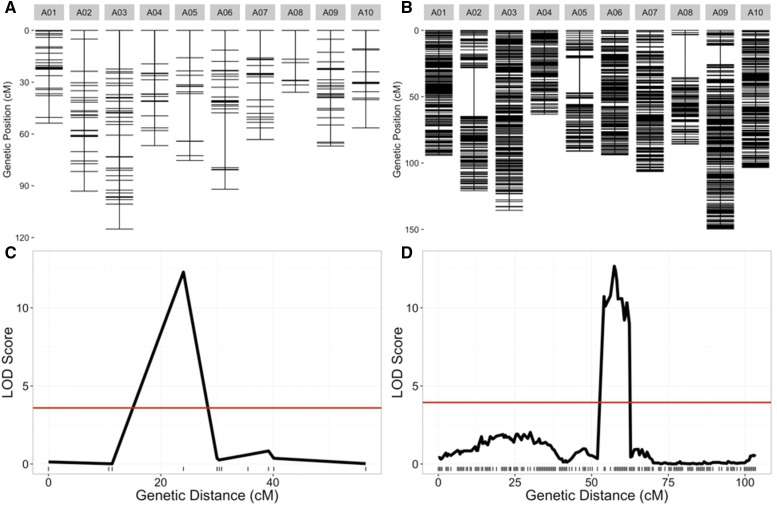
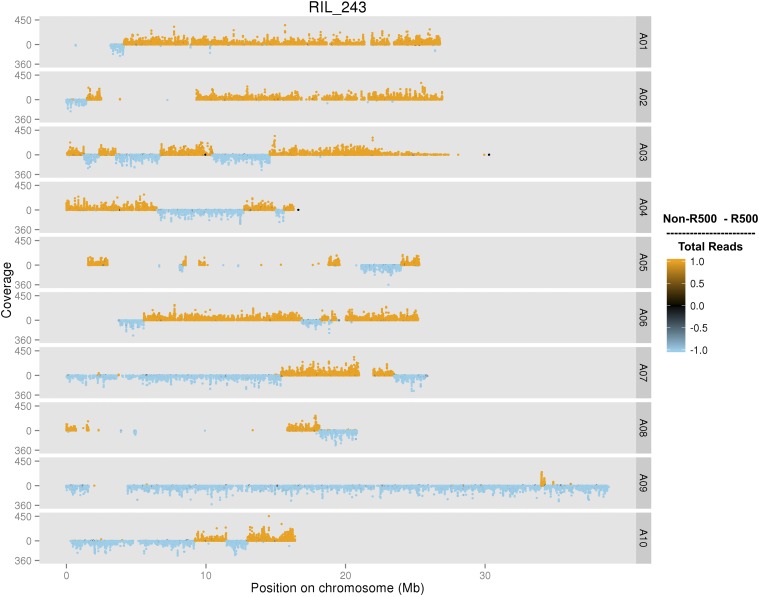
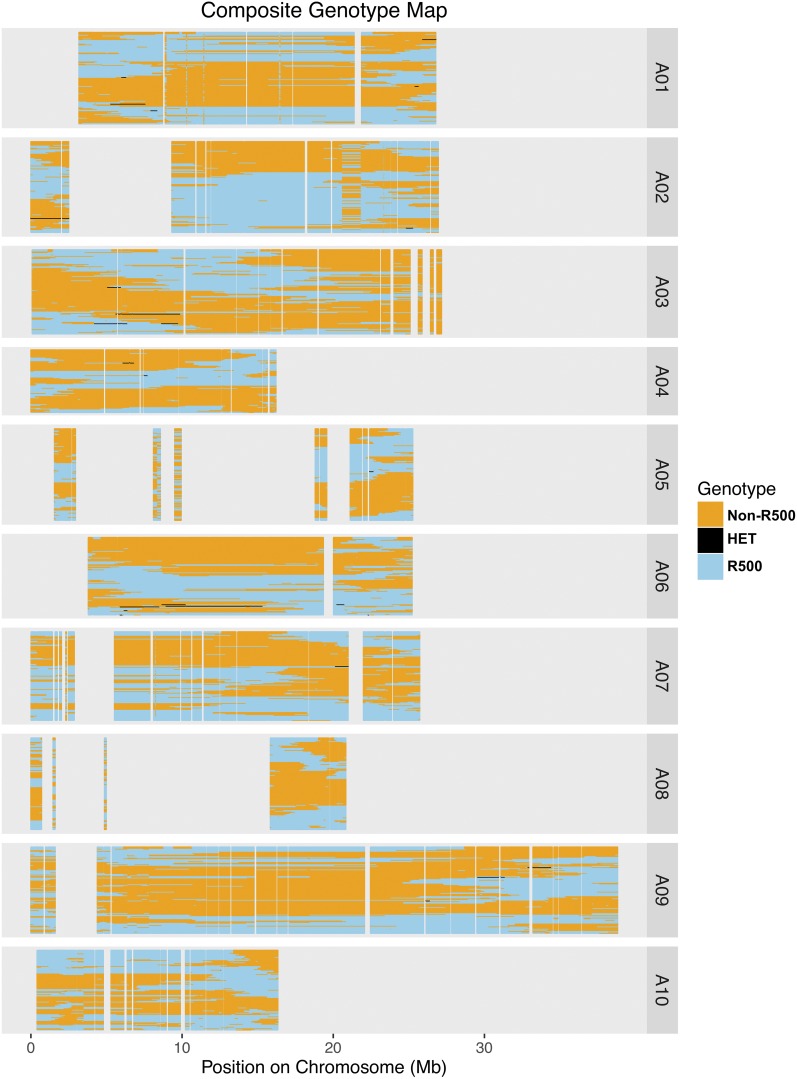
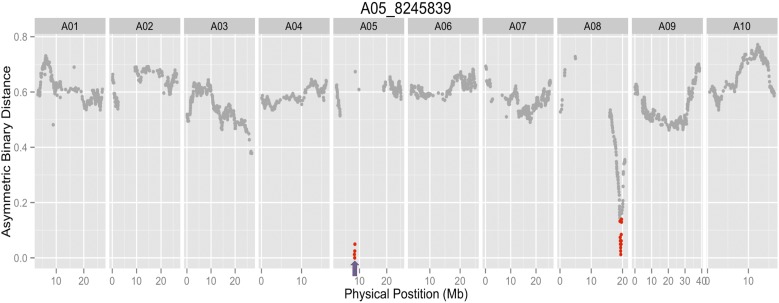
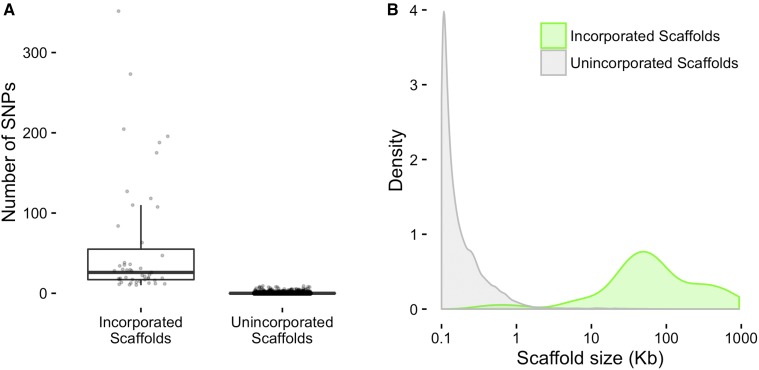
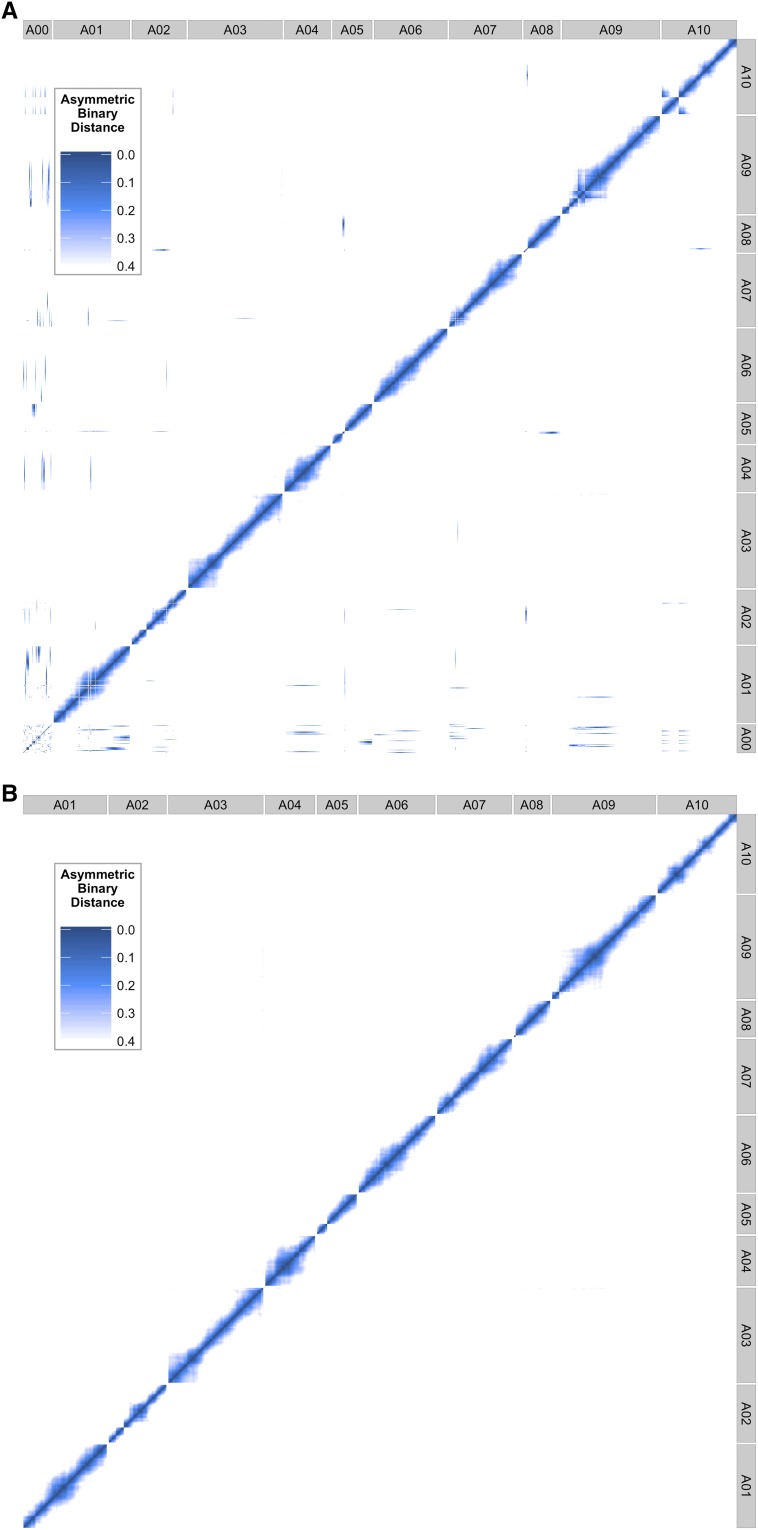
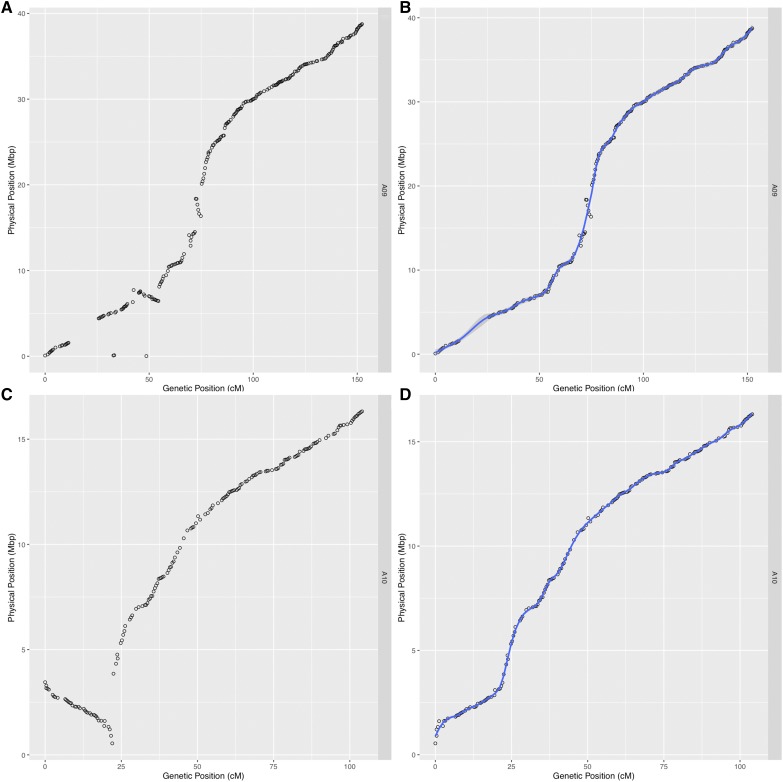
is a model species for agronomic, ecological, evolutionary, and translational studies. Here, we describe high-density SNP discovery and genetic map construction for a recombinant inbred line (RIL) population derived from field collected RNA sequencing (RNA-Seq) data. This high-density genotype data enables the detection and correction of putative genome misassemblies and accurate assignment of scaffold sequences to their likely genomic locations. These assembly improvements represent 7.1-8.0% of the annotated genome. We demonstrate how using this new resource leads to a significant improvement for QTL analysis over the current low-density genetic map. Improvements are achieved by the increased mapping resolution and by having known genomic coordinates to anchor the markers for candidate gene discovery. These new molecular resources and improvements in the genome annotation will benefit the Brassicaceae genomics community and may help guide other communities in fine-tuning genome annotations.
拟南芥是农艺学、生态学、进化生物学和转化研究的模式物种。在这里,我们描述了从田间收集的 RNA 测序(RNA-Seq)数据中衍生的重组自交系(RIL)群体的高密度 SNP 发现和遗传图谱构建。这种高密度基因型数据能够检测和校正可能的基因组组装错误,并将支架序列准确分配到其可能的基因组位置。这些组装改进占注释基因组的 7.1-8.0%。我们展示了如何利用这个新资源,通过增加图谱分辨率并为候选基因发现提供已知基因组坐标来锚定标记,从而显著提高 QTL 分析的准确性。这些新的分子资源和基因组注释的改进将使芸薹属基因组学社区受益,并可能有助于指导其他社区对基因组注释进行微调。