Wang Cheng, Peng Jingjin, Kuang Yanling, Zhang Jiaqiang, Dai Luming
The Second Department of Respiratory Medicine, The First Affiliated Hospital of Kunming Medical University, Kunming, Yunnan 650032, P.R. China.
Mol Med Rep. 2017 Aug;16(2):1147-1156. doi: 10.3892/mmr.2017.6758. Epub 2017 Jun 12.
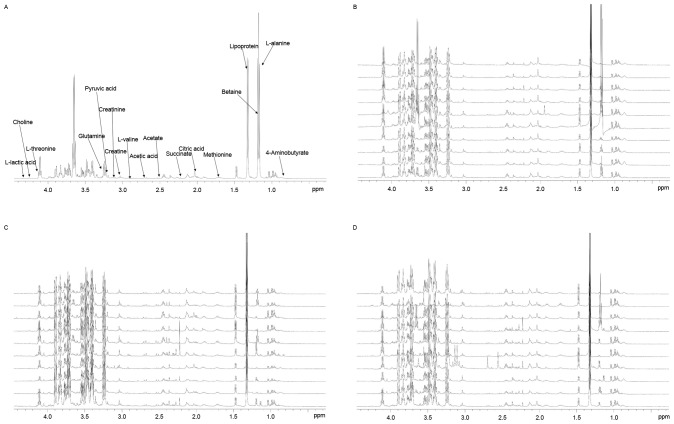
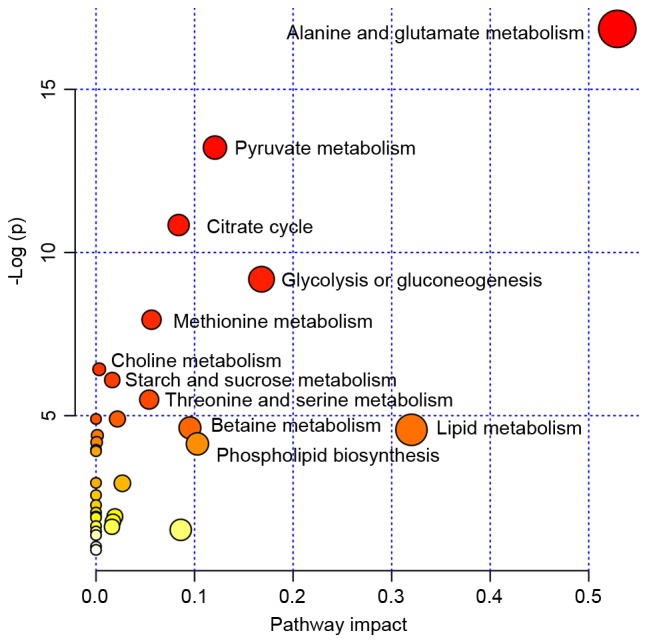
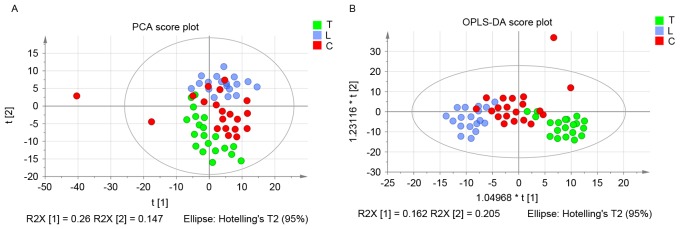
Pleural effusion is a common clinical manifestation with various causes. Current diagnostic and therapeutic methods have exhibited numerous limitations. By involving the analysis of dynamic changes in low molecular weight catabolites, metabolomics has been widely applied in various types of disease and have provided platforms to distinguish many novel biomarkers. However, to the best of our knowledge, there are few studies regarding the metabolic profiling for pleural effusion. In the current study, 58 pleural effusion samples were collected, among which 20 were malignant pleural effusions, 20 were tuberculous pleural effusions and 18 were transudative pleural effusions. The small molecule metabolite spectrums were obtained by adopting 1H nuclear magnetic resonance technology, and pattern‑recognition multi-variable statistical analysis was used to screen out different metabolites. One‑way analysis of variance, and Student‑Newman‑Keuls and the Kruskal‑Wallis test were adopted for statistical analysis. Over 400 metabolites were identified in the untargeted metabolomic analysis and 26 metabolites were identified as significantly different among tuberculous, malignant and transudative pleural effusions. These metabolites were predominantly involved in the metabolic pathways of amino acids metabolism, glycometabolism and lipid metabolism. Statistical analysis revealed that eight metabolites contributed to the distinction between the three groups: Tuberculous, malignant and transudative pleural effusion. In the current study, the feasibility of identifying small molecule biochemical profiles in different types of pleural effusion were investigated reveal novel biological insights into the underlying mechanisms. The results provide specific insights into the biology of tubercular, malignant and transudative pleural effusion and may offer novel strategies for the diagnosis and therapy of associated diseases, including tuberculosis, advanced lung cancer and congestive heart failure.
胸腔积液是一种常见的临床表现,病因多样。目前的诊断和治疗方法存在诸多局限性。代谢组学通过分析低分子量分解代谢物的动态变化,已广泛应用于各类疾病,并为鉴别许多新型生物标志物提供了平台。然而,据我们所知,关于胸腔积液代谢谱的研究较少。在本研究中,收集了58份胸腔积液样本,其中20份为恶性胸腔积液,20份为结核性胸腔积液,18份为漏出性胸腔积液。采用1H核磁共振技术获得小分子代谢物谱,并运用模式识别多变量统计分析筛选出不同的代谢物。采用单因素方差分析、Student-Newman-Keuls检验和Kruskal-Wallis检验进行统计分析。在非靶向代谢组学分析中鉴定出400多种代谢物,其中26种代谢物在结核性、恶性和漏出性胸腔积液中存在显著差异。这些代谢物主要参与氨基酸代谢、糖代谢和脂质代谢的代谢途径。统计分析表明,有8种代谢物有助于区分结核性、恶性和漏出性胸腔积液这三组。在本研究中,我们研究了识别不同类型胸腔积液中小分子生化谱的可行性,以揭示潜在机制的新生物学见解。研究结果为结核性、恶性和漏出性胸腔积液的生物学特性提供了具体见解,并可能为包括结核病、晚期肺癌和充血性心力衰竭在内的相关疾病的诊断和治疗提供新策略。