Liu Lian, Zhang Shao-Wu, Huang Yufei, Meng Jia
Key Laboratory of Information Fusion Technology of Ministry of Education, School of Automation, Northwestern Polytechnical University, Xi'an, 710072, China.
Department of Electrical and Computation Engineering, University of Texas at San Antonio, San Antonio, TX, 78230, USA.
BMC Bioinformatics. 2017 Aug 31;18(1):387. doi: 10.1186/s12859-017-1808-4.
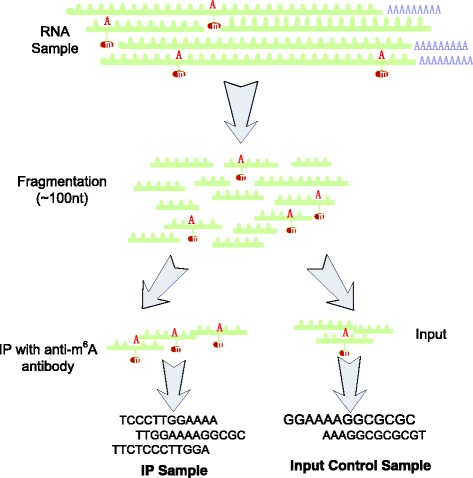
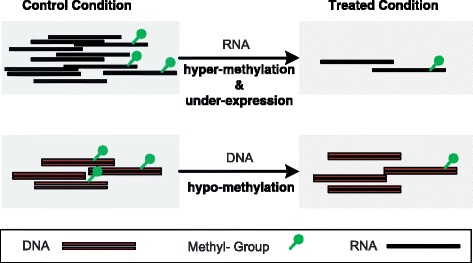
As a newly emerged research area, RNA epigenetics has drawn increasing attention recently for the participation of RNA methylation and other modifications in a number of crucial biological processes. Thanks to high throughput sequencing techniques, such as, MeRIP-Seq, transcriptome-wide RNA methylation profile is now available in the form of count-based data, with which it is often of interests to study the dynamics at epitranscriptomic layer. However, the sample size of RNA methylation experiment is usually very small due to its costs; and additionally, there usually exist a large number of genes whose methylation level cannot be accurately estimated due to their low expression level, making differential RNA methylation analysis a difficult task.
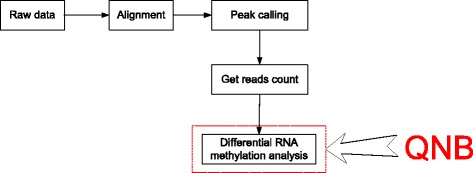
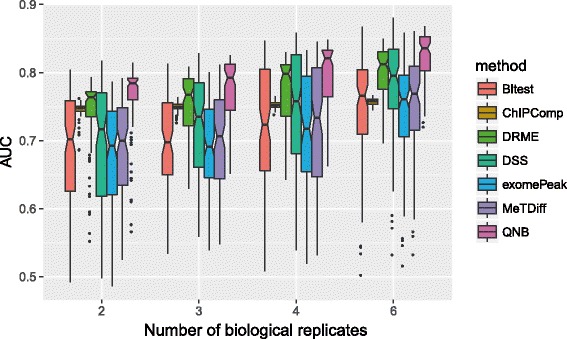
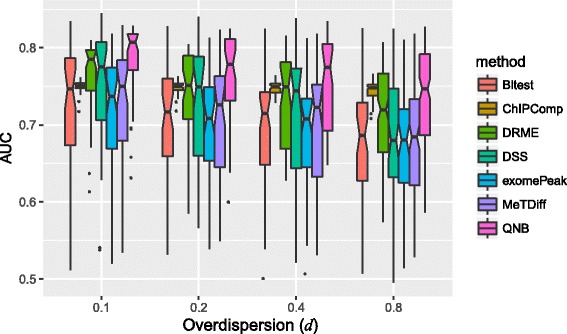
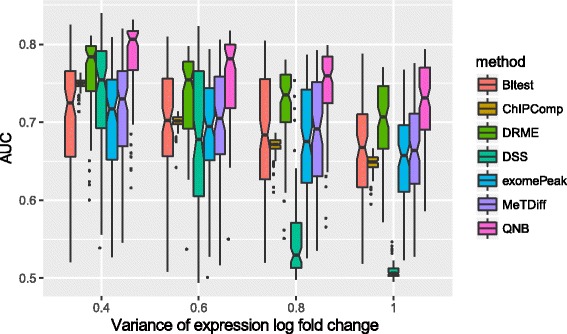
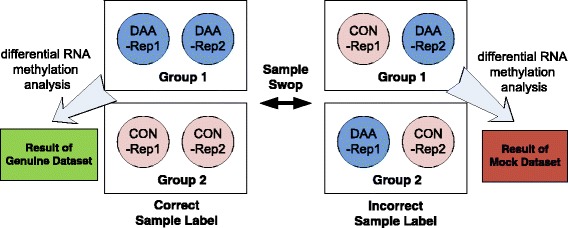
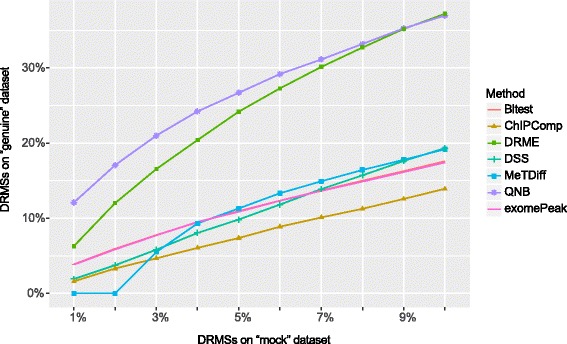
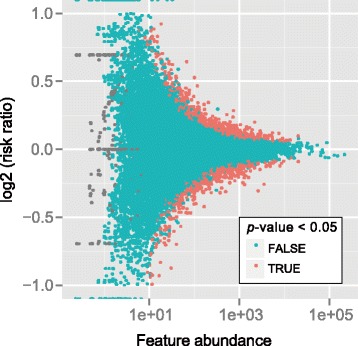
We present QNB, a statistical approach for differential RNA methylation analysis with count-based small-sample sequencing data. Compared with previous approaches such as DRME model based on a statistical test covering the IP samples only with 2 negative binomial distributions, QNB is based on 4 independent negative binomial distributions with their variances and means linked by local regressions, and in the way, the input control samples are also properly taken care of. In addition, different from DRME approach, which relies only the input control sample only for estimating the background, QNB uses a more robust estimator for gene expression by combining information from both input and IP samples, which could largely improve the testing performance for very lowly expressed genes.
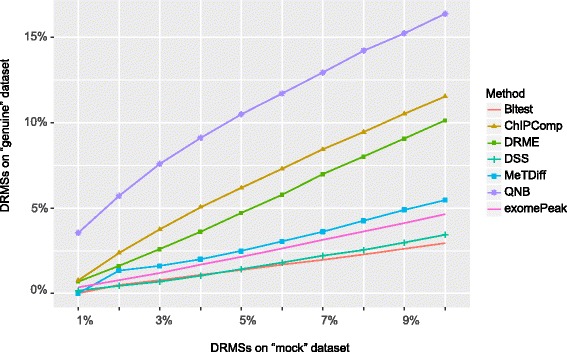
QNB showed improved performance on both simulated and real MeRIP-Seq datasets when compared with competing algorithms. And the QNB model is also applicable to other datasets related RNA modifications, including but not limited to RNA bisulfite sequencing, mA-Seq, Par-CLIP, RIP-Seq, etc.
作为一个新兴的研究领域,RNA表观遗传学最近因RNA甲基化和其他修饰参与许多关键生物学过程而受到越来越多的关注。得益于高通量测序技术,如MeRIP-Seq,现在可以以基于计数的数据形式获得全转录组范围的RNA甲基化图谱,利用这些数据研究表观转录组层面的动态变化常常很有意义。然而,由于成本原因,RNA甲基化实验的样本量通常非常小;此外,通常存在大量基因,由于其表达水平低,其甲基化水平无法准确估计,这使得差异RNA甲基化分析成为一项艰巨的任务。
我们提出了QNB,一种用于基于计数的小样本测序数据进行差异RNA甲基化分析的统计方法。与以前的方法,如基于仅用两个负二项分布覆盖IP样本的统计检验的DRME模型相比,QNB基于4个独立的负二项分布,其方差和均值通过局部回归联系起来,通过这种方式,输入对照样本也得到了妥善处理。此外,与仅依赖输入对照样本估计背景的DRME方法不同,QNB通过结合输入样本和IP样本的信息,对基因表达使用了更稳健的估计器,这可以大大提高对极低表达基因的检测性能。
与竞争算法相比,QNB在模拟和真实的MeRIP-Seq数据集上均表现出更好的性能。并且QNB模型也适用于其他与RNA修饰相关的数据集,包括但不限于RNA亚硫酸氢盐测序、mA-Seq、Par-CLIP、RIP-Seq等。