Department of Botany and Plant Sciences, University of California, Riverside, USA.
Sci Rep. 2017 Oct 20;7(1):13678. doi: 10.1038/s41598-017-14070-z.
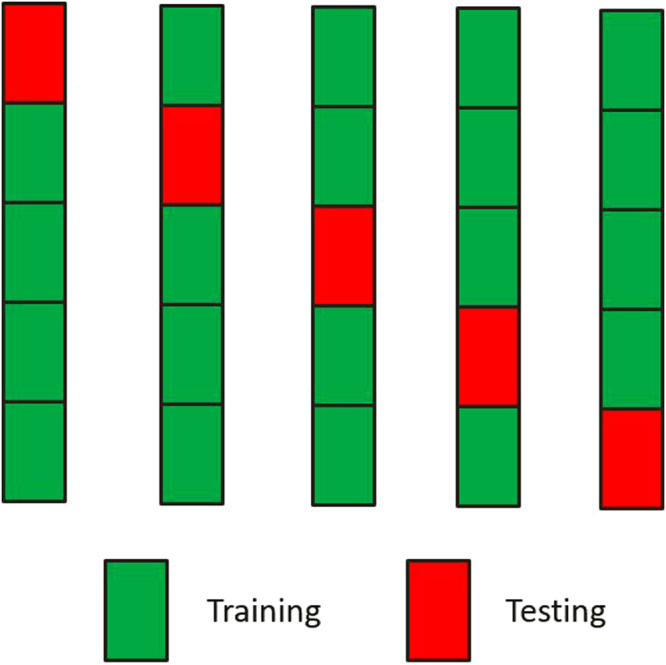
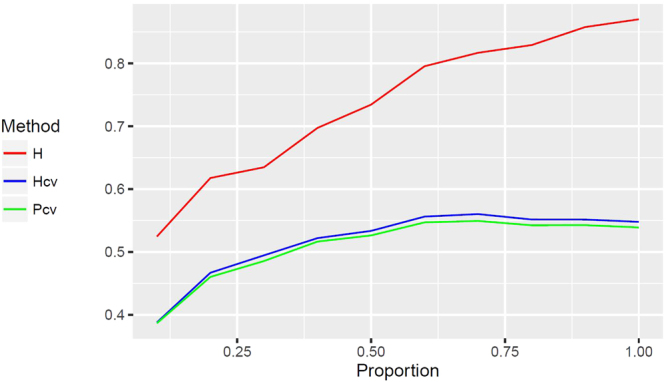
In genomic selection (GS), all the markers across the entire genome are used to conduct marker-assisted selection such that each quantitative trait locus of complex trait is in linkage disequilibrium with at least one marker. Although GS improves estimated breeding values and genetic gain, in most GS models genetic variance is estimated from training samples with many trait-irrelevant markers, which leads to severe overfitting in the calculation of trait heritability. In this study, we demonstrated overfitting heritability due to the inclusion of trait-irrelevant markers using a series of simulations, and such overfitting can be effectively controlled by cross validation experiment. In the proposed method, the genetic variance is simply the variance of the genetic values predicted through cross validation, the residual variance is the variance of the differences between the observed phenotypic values and the predicted genetic values, and these two resultant variance components are used for calculating the unbiased heritability. We also demonstrated that the heritability calculated through cross validation is equivalent to trait predictability, which objectively reflects the applicability of the GS models. The proposed method can be implemented with the Mixed Procedure in SAS or with our R package "GSMX" which is publically available at https://cran.r-project.org/web/packages/GSMX/index.html .
在基因组选择(GS)中,使用整个基因组中的所有标记来进行标记辅助选择,使得复杂性状的每个数量性状位点都与至少一个标记处于连锁不平衡状态。尽管 GS 提高了估计的育种值和遗传增益,但在大多数 GS 模型中,遗传方差是从包含许多与性状无关的标记的训练样本中估计的,这导致在计算性状遗传力时严重过度拟合。在这项研究中,我们通过一系列模拟演示了由于包含与性状无关的标记而导致的遗传力过度拟合,并且可以通过交叉验证实验有效地控制这种过度拟合。在所提出的方法中,遗传方差只是通过交叉验证预测的遗传值的方差,剩余方差是观察到的表型值和预测的遗传值之间的差异的方差,这两个结果方差分量用于计算无偏遗传力。我们还证明了通过交叉验证计算的遗传力等同于性状可预测性,这客观地反映了 GS 模型的适用性。该方法可以通过 SAS 中的混合过程或我们的 R 包“GSMX”实现,该 R 包可在 https://cran.r-project.org/web/packages/GSMX/index.html 上公开获得。