Innovative Drug Research and Bioinformatics Group, School of Pharmaceutical Sciences and Collaborative Innovation Center for Brain Science, Chongqing University, Chongqing 401331, China.
Innovative Drug Research and Bioinformatics Group, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, China.
Int J Mol Sci. 2018 Jan 8;19(1):183. doi: 10.3390/ijms19010183.
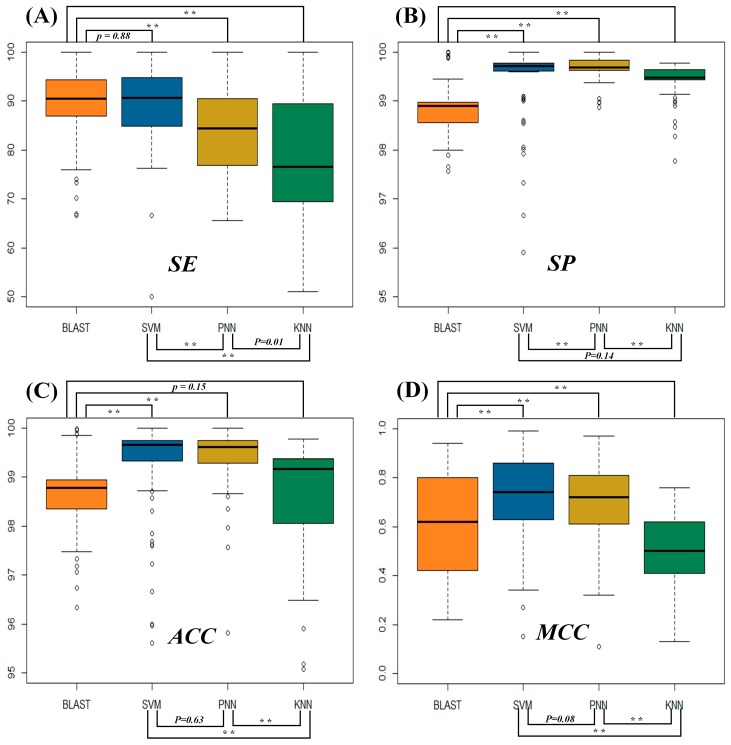
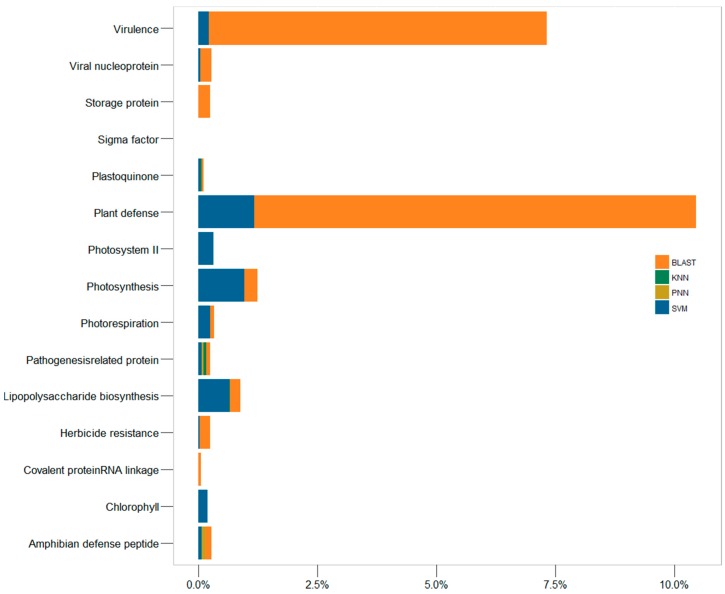
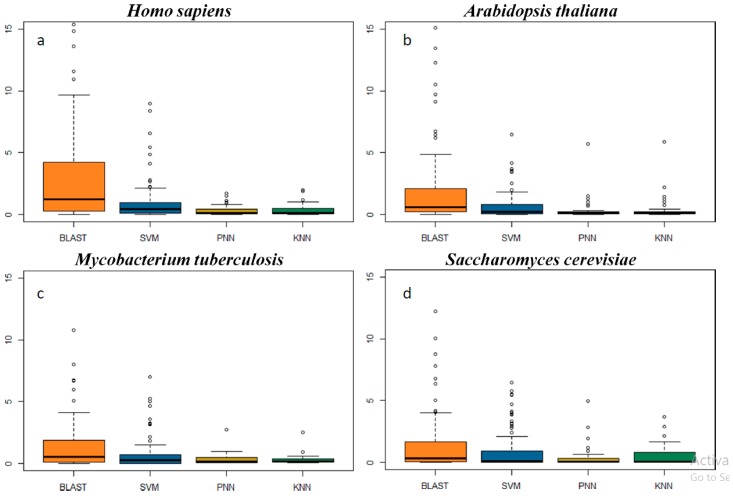
The function of a protein is of great interest in the cutting-edge research of biological mechanisms, disease development and drug/target discovery. Besides experimental explorations, a variety of computational methods have been designed to predict protein function. Among these in silico methods, the prediction of BLAST is based on protein sequence similarity, while that of machine learning is also based on the sequence, but without the consideration of their similarity. This unique characteristic of machine learning makes it a good complement to BLAST and many other approaches in predicting the function of remotely relevant proteins and the homologous proteins of distinct function. However, the identification accuracies of these in silico methods and their false discovery rate have not yet been assessed so far, which greatly limits the usage of these algorithms. Herein, a comprehensive comparison of the performances among four popular prediction algorithms (BLAST, SVM, PNN and KNN) was conducted. In particular, the performance of these methods was systematically assessed by four standard statistical indexes based on the independent test datasets of 93 functional protein families defined by UniProtKB keywords. Moreover, the false discovery rates of these algorithms were evaluated by scanning the genomes of four representative model organisms (, , and ). As a result, the substantially higher sensitivity of SVM and BLAST was observed compared with that of PNN and KNN. However, the machine learning algorithms (PNN, KNN and SVM) were found capable of substantially reducing the false discovery rate (SVM < PNN < KNN). In sum, this study comprehensively assessed the performance of four popular algorithms applied to protein function prediction, which could facilitate the selection of the most appropriate method in the related biomedical research.
蛋白质的功能是生物机制、疾病发展和药物/靶点发现的前沿研究中非常感兴趣的问题。除了实验探索外,还设计了各种计算方法来预测蛋白质功能。在这些计算方法中,BLAST 的预测基于蛋白质序列相似性,而机器学习的预测也是基于序列,但不考虑它们的相似性。机器学习的这个独特特征使其成为 BLAST 和许多其他方法的良好补充,可以预测远程相关蛋白质和功能不同的同源蛋白质的功能。然而,到目前为止,这些计算方法的识别精度及其假发现率尚未得到评估,这极大地限制了这些算法的使用。在此,我们对四种流行的预测算法(BLAST、SVM、PNN 和 KNN)的性能进行了全面比较。特别是,基于 UniProtKB 关键字定义的 93 个功能蛋白家族的独立测试数据集,使用四个标准统计指标系统地评估了这些方法的性能。此外,还通过扫描四个代表性模式生物(、、和)的基因组来评估这些算法的假发现率。结果表明,SVM 和 BLAST 的灵敏度明显高于 PNN 和 KNN。然而,机器学习算法(PNN、KNN 和 SVM)被发现能够大大降低假发现率(SVM < PNN < KNN)。总之,本研究全面评估了四种流行算法在蛋白质功能预测中的性能,这有助于在相关的生物医学研究中选择最合适的方法。