Institut des Sciences de l'Evolution (ISEM), UMR 5554, CNRS, IRD, EPHE, Université de Montpellier, Montpellier, France.
Sorbonne Université, CNRS, Institut de Biologie Paris-Seine (IBPS), Evolution Paris-Seine (UMR7138), Case 05, 7 Quai St Bernard, 75005, Paris, France.
BMC Biol. 2018 Mar 5;16(1):28. doi: 10.1186/s12915-018-0486-7.
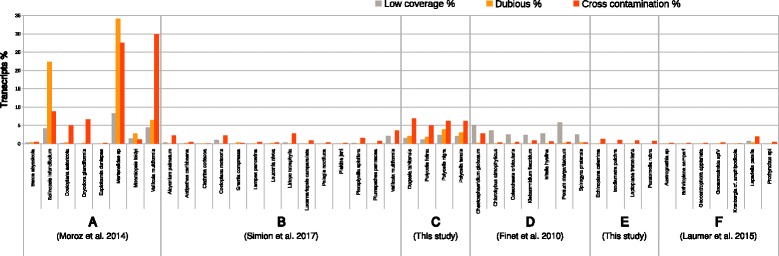
Multiple RNA samples are frequently processed together and often mixed before multiplex sequencing in the same sequencing run. While different samples can be separated post sequencing using sample barcodes, the possibility of cross contamination between biological samples from different species that have been processed or sequenced in parallel has the potential to be extremely deleterious for downstream analyses.
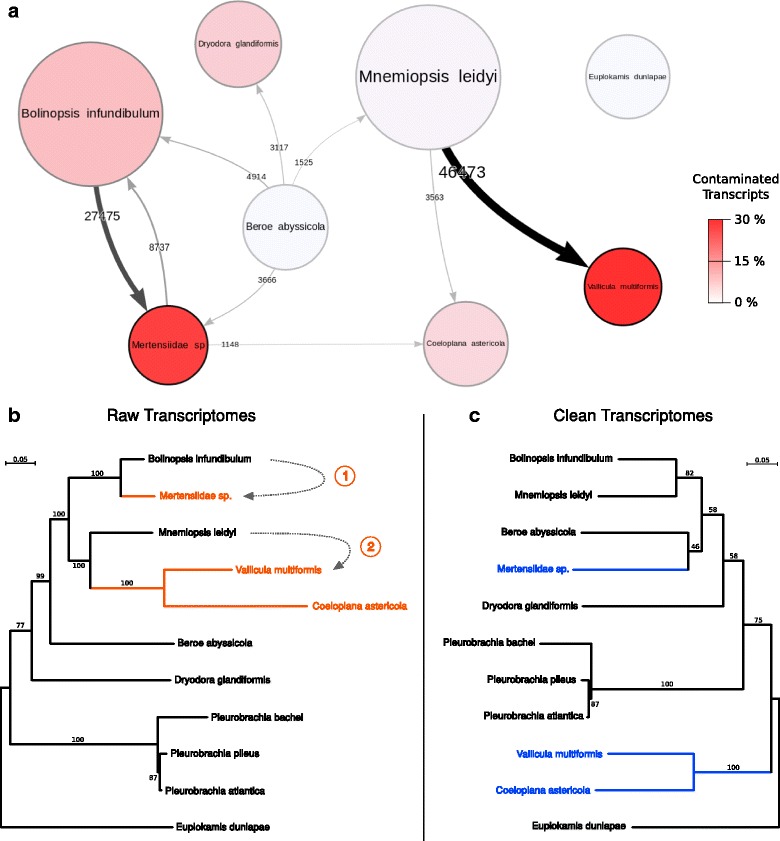
We present CroCo, a software package for identifying and removing such cross contaminants from assembled transcriptomes. Using multiple, recently published sequence datasets, we show that cross contamination is consistently present at varying levels in real data. Using real and simulated data, we demonstrate that CroCo detects contaminants efficiently and correctly. Using a real example from a molecular phylogenetic dataset, we show that contaminants, if not eliminated, can have a decisive, deleterious impact on downstream comparative analyses.
Cross contamination is pervasive in new and published datasets and, if undetected, can have serious deleterious effects on downstream analyses. CroCo is a database-independent, multi-platform tool, designed for ease of use, that efficiently and accurately detects and removes cross contamination in assembled transcriptomes to avoid these problems. We suggest that the use of CroCo should become a standard cleaning step when processing multiple samples for transcriptome sequencing.
在同一测序运行中,通常会将多个 RNA 样本一起处理并经常混合进行多重测序。虽然可以在测序后使用样本条形码将不同的样本分开,但来自不同物种的生物样本在并行处理或测序过程中发生交叉污染的可能性,会对下游分析造成极大的危害。
我们提出了 CroCo,这是一种用于从组装转录组中识别和去除这种交叉污染的软件包。使用多个最近发表的序列数据集,我们表明交叉污染在真实数据中以不同的水平持续存在。使用真实和模拟数据,我们证明了 CroCo 能够有效地和正确地检测到污染物。使用来自分子系统发育数据集的真实示例,我们表明,如果不消除污染物,它们会对下游的比较分析产生决定性的有害影响。
交叉污染在新的和已发表的数据集中普遍存在,如果未被发现,会对下游分析产生严重的有害影响。CroCo 是一个独立于数据库、多平台的工具,旨在易于使用,能够有效地和准确地检测和去除组装转录组中的交叉污染,从而避免这些问题。我们建议,在进行多个样本的转录组测序时,应将使用 CroCo 作为标准的清理步骤。