National Engineering Laboratory for Animal Breeding, Key Laboratory of Animal Genetics, Breeding and Reproduction, Ministry of Agriculture, College of Animal Science and Technology, China Agricultural University, Beijing, 100193, China.
Animal Biosciences, International Livestock Research Institute, Nairobi, 00100, Kenya.
Genet Sel Evol. 2018 Mar 26;50(1):12. doi: 10.1186/s12711-018-0383-0.
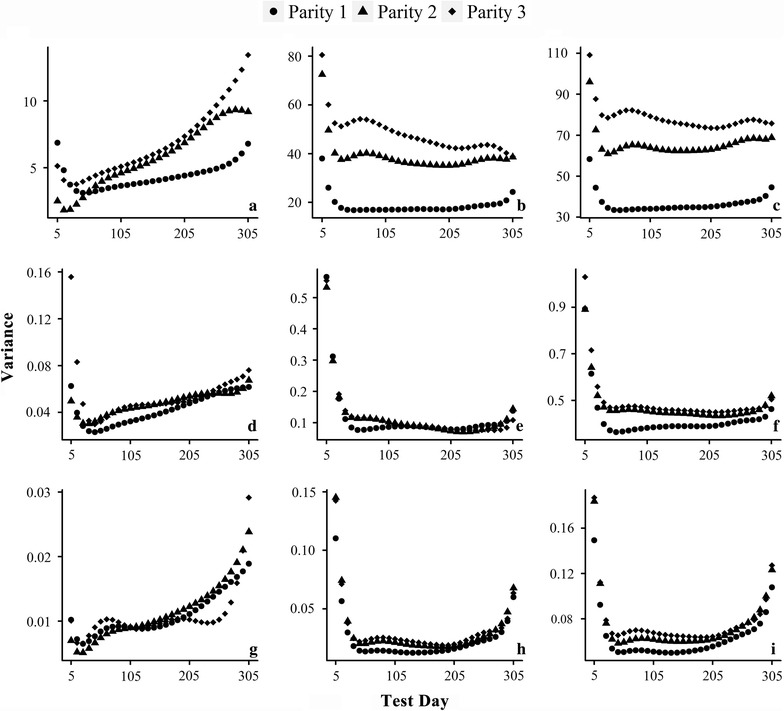
Pseudo-phenotypes, such as 305-day yields, estimated breeding values or deregressed proofs, are usually used as response variables for genome-wide association studies (GWAS) of milk production traits in dairy cattle. Computational inefficiency challenges the direct use of test-day records for longitudinal GWAS with large datasets.
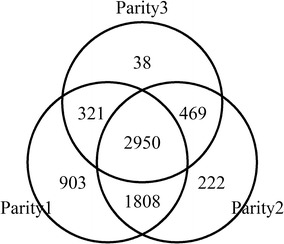
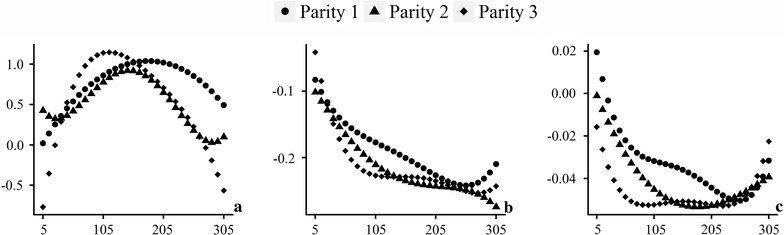
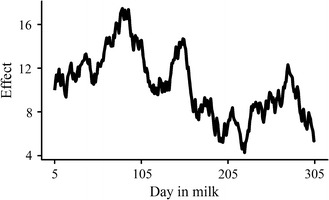
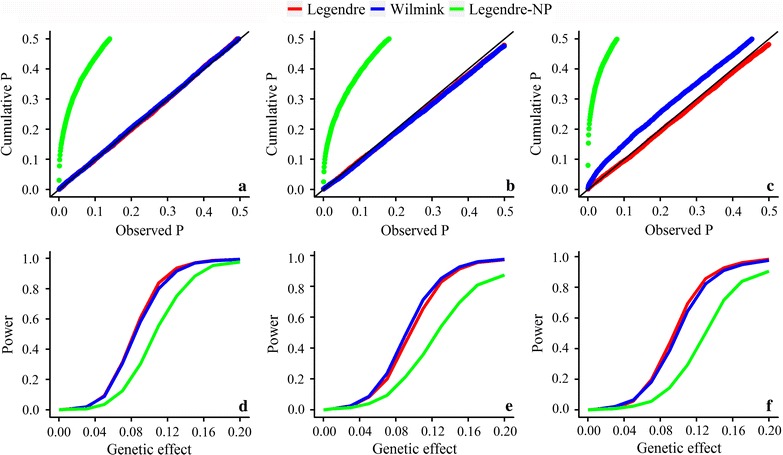
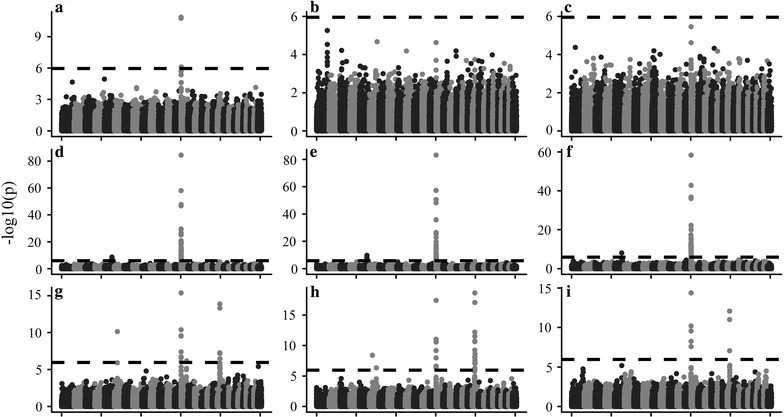
We propose a rapid longitudinal GWAS method that is based on a random regression model. Our method uses Eigen decomposition of the phenotypic covariance matrix to rotate the data, thereby transforming the complex mixed linear model into weighted least squares analysis. We performed a simulation study that showed that our method can control type I errors well and has higher power than a longitudinal GWAS method that does not include time-varied additive genetic effects. We also applied our method to the analysis of milk production traits in the first three parities of 6711 Chinese Holstein cows. The analysis for each trait was completed within 1 day with known variances. In total, we located 84 significant single nucleotide polymorphisms (SNPs) of which 65 were within previously reported quantitative trait loci (QTL) regions.
Our rapid method can control type I errors in the analysis of longitudinal data and can be applied to other longitudinal traits. We detected QTL that were for the most part similar to those reported in a previous study in Chinese Holstein. Moreover, six additional SNPs for fat percentage and 13 SNPs for protein percentage were identified by our method. These additional 19 SNPs could be new candidate quantitative trait nucleotides for milk production traits in Chinese Holstein.
在奶牛生产性状的全基因组关联研究(GWAS)中,通常使用 305 天产奶量、估计育种值或去回归验证等伪表型作为响应变量。计算效率低下的问题挑战了直接使用测试日记录进行大型数据集的纵向 GWAS。
我们提出了一种基于随机回归模型的快速纵向 GWAS 方法。我们的方法使用表型协方差矩阵的特征分解来旋转数据,从而将复杂的混合线性模型转换为加权最小二乘分析。我们进行了一项模拟研究,表明我们的方法可以很好地控制 I 型错误,并且比不包括时变加性遗传效应的纵向 GWAS 方法具有更高的功效。我们还将我们的方法应用于 6711 头中国荷斯坦奶牛前三个泌乳期的产奶性状分析。对于每个性状的分析在已知方差的情况下可以在 1 天内完成。总共定位到 84 个显著的单核苷酸多态性(SNP),其中 65 个位于先前报道的数量性状位点(QTL)区域内。
我们的快速方法可以控制纵向数据分析中的 I 型错误,并且可以应用于其他纵向性状。我们检测到的 QTL 在很大程度上与之前在中国荷斯坦牛中报道的 QTL 相似。此外,我们的方法还鉴定了脂肪百分率的 6 个额外 SNP 和蛋白质百分率的 13 个 SNP。这些额外的 19 个 SNP 可能是中国荷斯坦牛产奶性状的新候选数量性状核苷酸。