Department of Computer Science, University of Reading, Reading, UK.
BMC Bioinformatics. 2018 Apr 11;19(1):127. doi: 10.1186/s12859-018-2125-2.
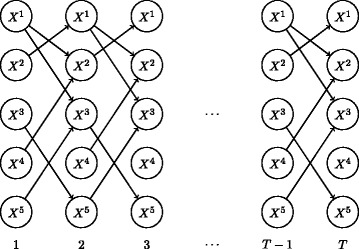
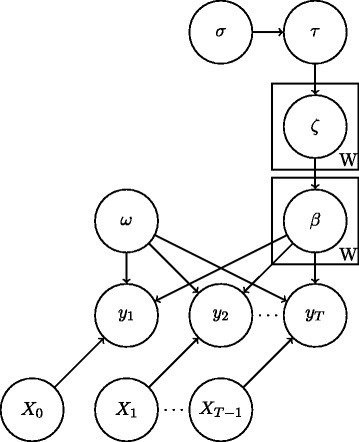
Inference of gene regulatory network structures from RNA-Seq data is challenging due to the nature of the data, as measurements take the form of counts of reads mapped to a given gene. Here we present a model for RNA-Seq time series data that applies a negative binomial distribution for the observations, and uses sparse regression with a horseshoe prior to learn a dynamic Bayesian network of interactions between genes. We use a variational inference scheme to learn approximate posterior distributions for the model parameters.
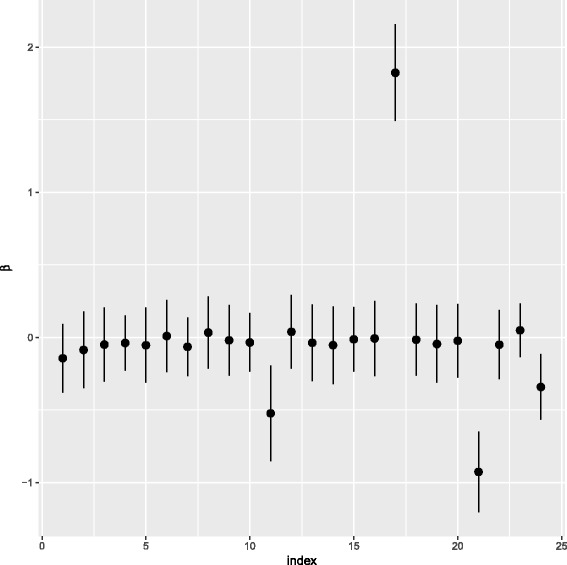
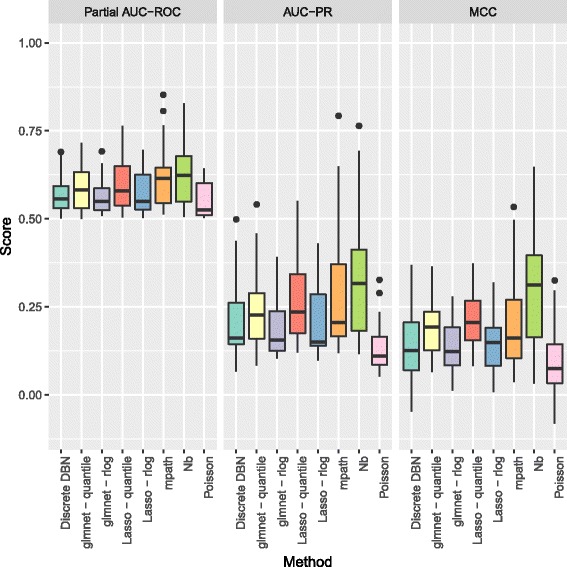
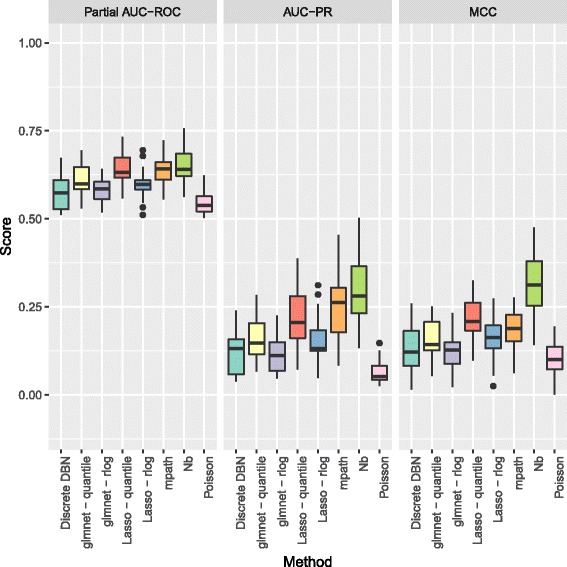
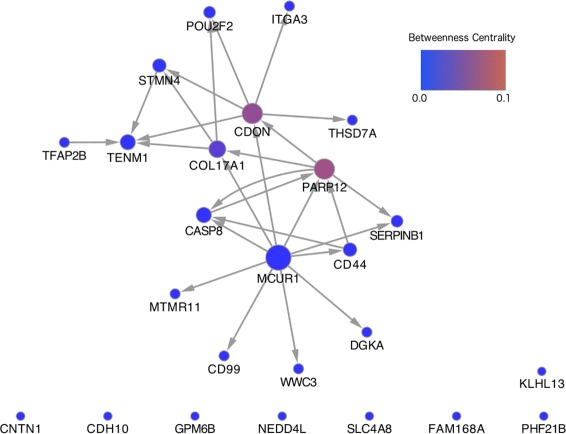
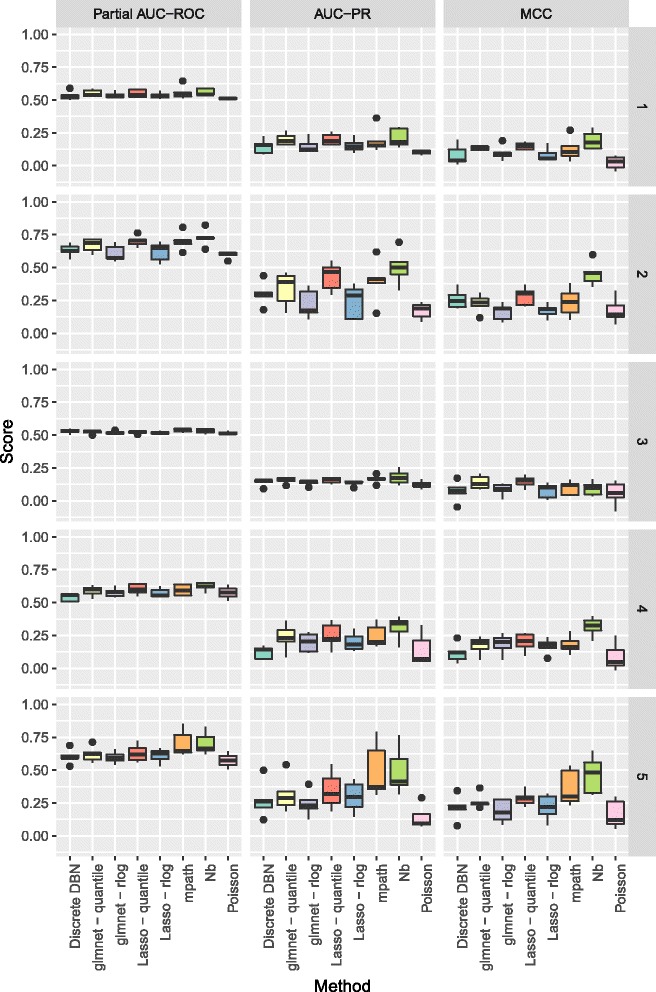
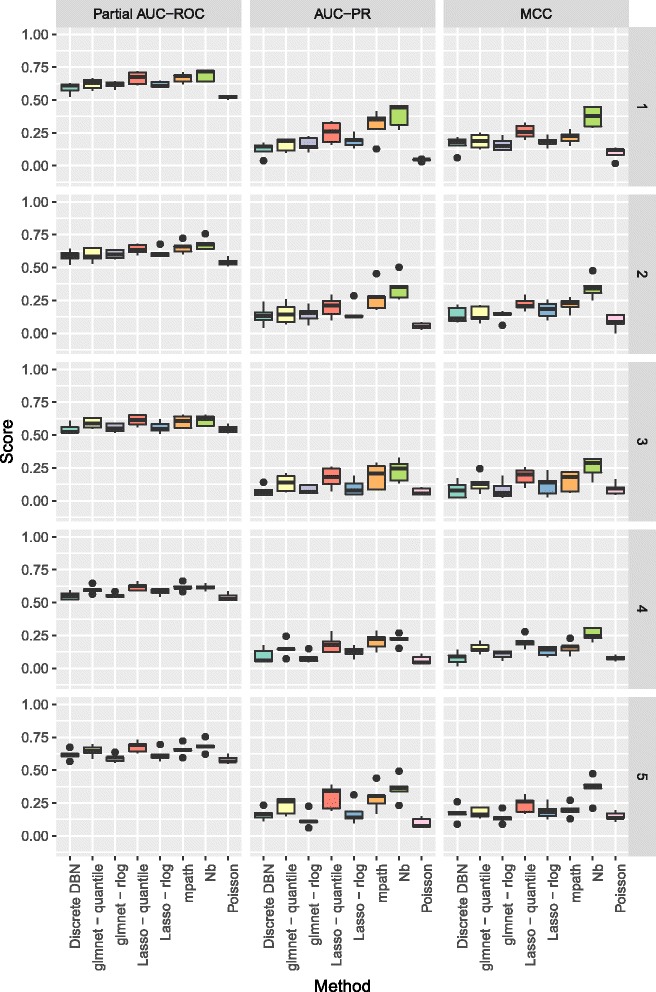
The methodology is benchmarked on synthetic data designed to replicate the distribution of real world RNA-Seq data. We compare our method to other sparse regression approaches and find improved performance in learning directed networks. We demonstrate an application of our method to a publicly available human neuronal stem cell differentiation RNA-Seq time series data set to infer the underlying network structure.
Our method is able to improve performance on synthetic data by explicitly modelling the statistical distribution of the data when learning networks from RNA-Seq time series. Applying approximate inference techniques we can learn network structures quickly with only moderate computing resources.
由于数据的性质,从 RNA-Seq 数据中推断基因调控网络结构具有挑战性,因为测量采用的是映射到给定基因的读取计数的形式。在这里,我们提出了一个适用于 RNA-Seq 时间序列数据的模型,该模型对观测值应用负二项分布,并使用具有马氏先验的稀疏回归来学习基因之间相互作用的动态贝叶斯网络。我们使用变分推理方案来学习模型参数的近似后验分布。
该方法在设计用于复制真实 RNA-Seq 数据分布的合成数据上进行了基准测试。我们将我们的方法与其他稀疏回归方法进行了比较,并发现它在学习有向网络方面的性能有所提高。我们展示了我们的方法在公开可用的人类神经元干细胞分化 RNA-Seq 时间序列数据集上的应用,以推断潜在的网络结构。
我们的方法通过在从 RNA-Seq 时间序列学习网络时显式地对数据的统计分布进行建模,从而能够在合成数据上提高性能。通过应用近似推理技术,我们可以在仅使用适度计算资源的情况下快速学习网络结构。