Yao Zhenpeng, Kim Soo, He Jiangang, Hegde Vinay I, Wolverton Chris
Department of Materials Science and Engineering, Northwestern University, 2220 Campus Drive, Evanston, IL 60208, USA.
Sci Adv. 2018 May 18;4(5):eaao6754. doi: 10.1126/sciadv.aao6754. eCollection 2018 May.
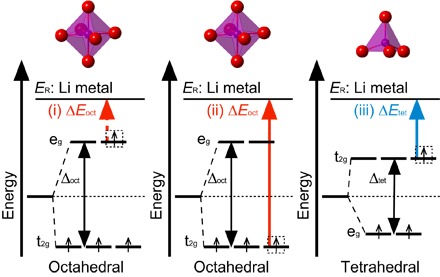
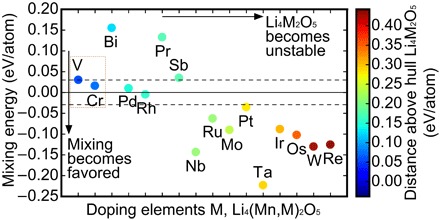
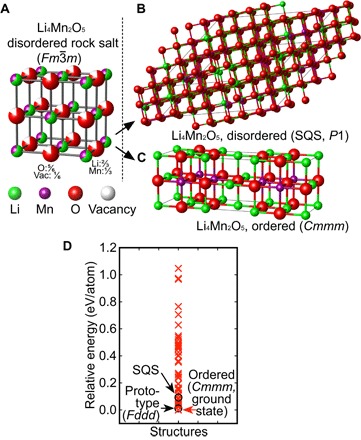
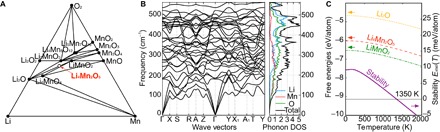
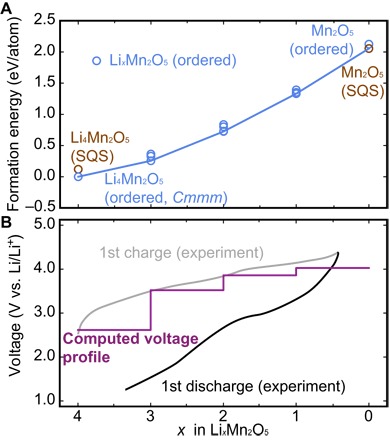
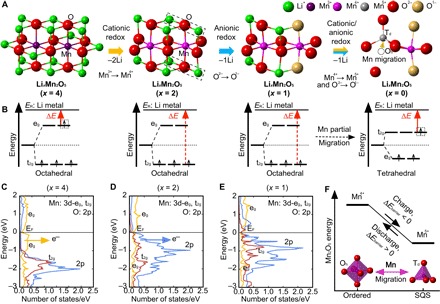
Significant research effort has focused on improving the specific energy of lithium-ion batteries for emerging applications, such as electric vehicles. Recently, a rock salt-type LiMnO cathode material with a large discharge capacity (~350 mA·hour g) was discovered. However, a full structural model of LiMnO and its corresponding phase transformations, as well as the atomistic origins of the high capacity, warrants further investigation. We use first-principles density functional theory (DFT) calculations to investigate both the disordered rock salt-type LiMnO structure and the ordered ground-state structure. The ionic ordering in the ground-state structure is determined via a DFT-based enumeration method. We use both the ordered and disordered structures to interrogate the delithiation process and find that it occurs via a three-step reaction pathway involving the complex interplay of cation and anion redox reactions: (i) an initial metal oxidation, Mn→Mn (Li MnO, 4 > > 2); (ii) followed by anion oxidation, O→O (2 > > 1); and (iii) finally, further metal oxidation, Mn→Mn (1 > > 0). This final step is concomitant with the Mn migration from the original octahedral site to the adjacent tetrahedral site, introducing a kinetic barrier to reversible charge/discharge cycles. Armed with this knowledge of the charging process, we use high-throughput DFT calculations to study metal mixing in this compound, screening potential new materials for stability and kinetic reversibility. We predict that mixing with M = V and Cr in Li(Mn,M)O will produce new stable compounds with substantially improved electrochemical properties.
大量的研究工作致力于提高锂离子电池的比能量,以满足诸如电动汽车等新兴应用的需求。最近,发现了一种具有大放电容量(约350 mA·小时/克)的岩盐型LiMnO阴极材料。然而,LiMnO的完整结构模型及其相应的相变,以及高容量的原子起源,仍有待进一步研究。我们使用第一性原理密度泛函理论(DFT)计算来研究无序岩盐型LiMnO结构和有序基态结构。基态结构中的离子有序性通过基于DFT的枚举方法确定。我们使用有序和无序结构来研究脱锂过程,发现它通过一个三步反应途径发生,涉及阳离子和阴离子氧化还原反应的复杂相互作用:(i)初始金属氧化,Mn→Mn(Li MnO,4 > > 2);(ii)随后是阴离子氧化,O→O(2 > > 1);(iii)最后,进一步的金属氧化,Mn→Mn(1 > > 0)。这最后一步伴随着Mn从原始八面体位置迁移到相邻四面体位置,给可逆充电/放电循环带来了动力学障碍。基于对充电过程的这一认识,我们使用高通量DFT计算来研究该化合物中的金属混合,筛选具有稳定性和动力学可逆性的潜在新材料。我们预测,在Li(Mn,M)O中与M = V和Cr混合将产生具有显著改善的电化学性能的新稳定化合物。