Iuliano Antonella, Occhipinti Annalisa, Angelini Claudia, De Feis Italia, Liò Pietro
Istituto per le Applicazioni del Calcolo "Mauro Picone", Consiglio Nazionale delle Ricerche, Naples, Italy.
Telethon Institute of Genetics and Medicine, Pozzuoli, Italy.
Front Genet. 2018 Jun 14;9:206. doi: 10.3389/fgene.2018.00206. eCollection 2018.
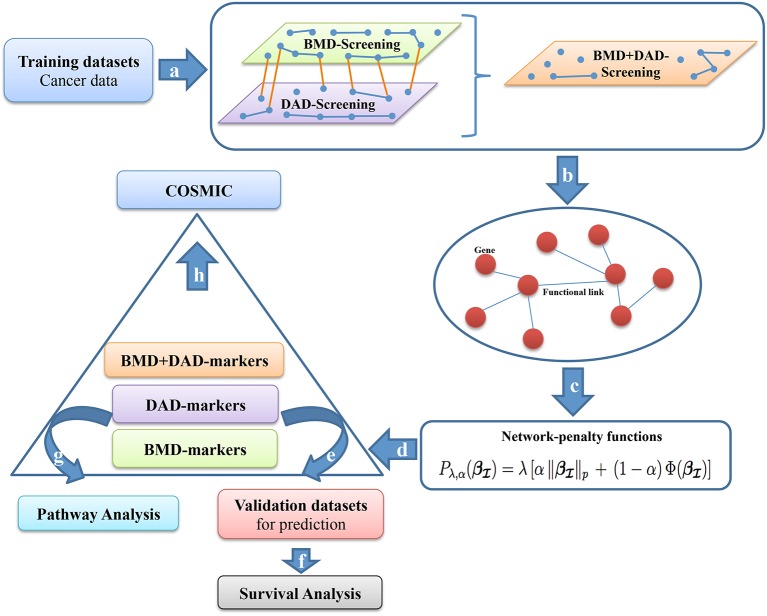
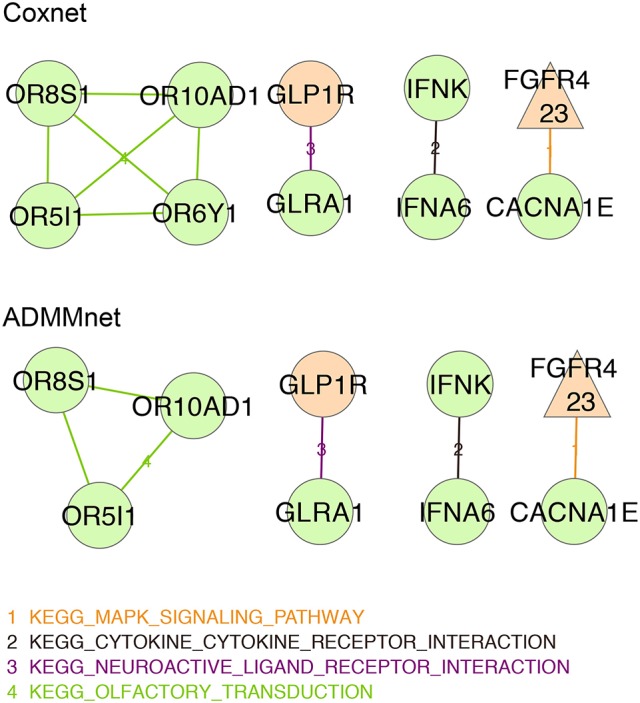
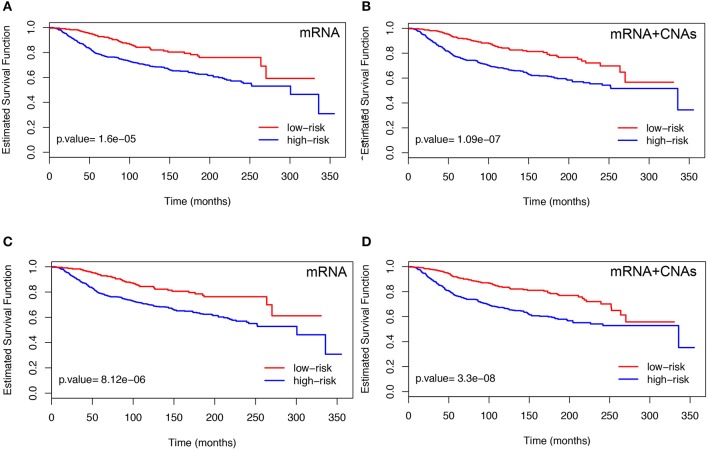
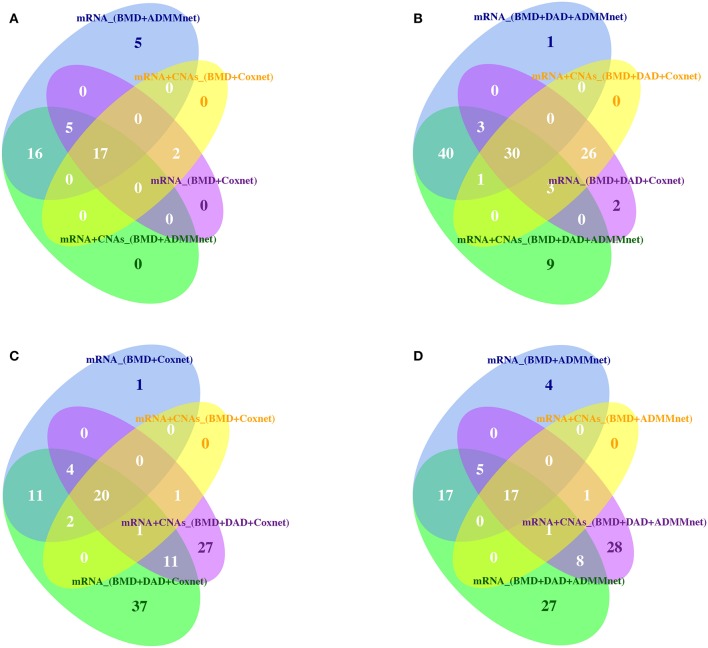
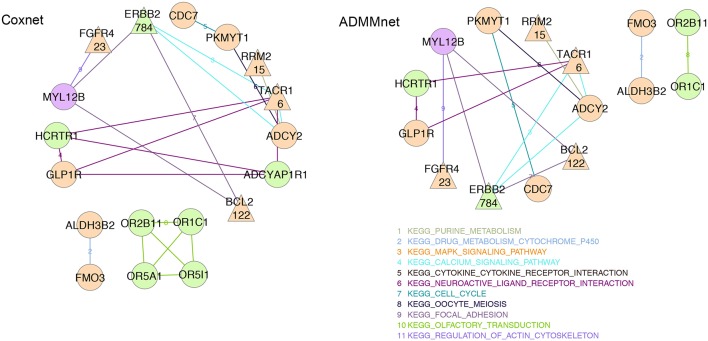
Breast cancer is one of the most common invasive tumors causing high mortality among women. It is characterized by high heterogeneity regarding its biological and clinical characteristics. Several high-throughput assays have been used to collect genome-wide information for many patients in large collaborative studies. This knowledge has improved our understanding of its biology and led to new methods of diagnosing and treating the disease. In particular, system biology has become a valid approach to obtain better insights into breast cancer biological mechanisms. A crucial component of current research lies in identifying novel biomarkers that can be predictive for breast cancer patient prognosis on the basis of the molecular signature of the tumor sample. However, the high dimension and low sample size of data greatly increase the difficulty of cancer survival analysis demanding for the development of statistical methods. In this work, we propose novel screening-network methods that predict patient survival outcome by screening key survival-related genes and we assess the capability of the proposed approaches using METABRIC dataset. In particular, we first identify a subset of genes by using variable screening techniques on gene expression data. Then, we perform Cox regression analysis by incorporating network information associated with the selected subset of genes. The novelty of this work consists in the improved prediction of survival responses due to the different types of screenings (i.e., a biomedical-driven, data-driven and a combination of the two) before building the network-penalized model. Indeed, the combination of the two screening approaches allows us to use the available biological knowledge on breast cancer and complement it with additional information emerging from the data used for the analysis. Moreover, we also illustrate how to extend the proposed approaches to integrate an additional omic layer, such as copy number aberrations, and we show that such strategies can further improve our prediction capabilities. In conclusion, our approaches allow to discriminate patients in high-and low-risk groups using few potential biomarkers and therefore, can help clinicians to provide more precise prognoses and to facilitate the subsequent clinical management of patients at risk of disease.
乳腺癌是导致女性高死亡率的最常见侵袭性肿瘤之一。其生物学和临床特征具有高度异质性。在大型合作研究中,已使用多种高通量检测方法为众多患者收集全基因组信息。这些知识增进了我们对其生物学的理解,并催生了诊断和治疗该疾病的新方法。特别是,系统生物学已成为深入了解乳腺癌生物学机制的有效途径。当前研究的一个关键组成部分在于识别新型生物标志物,这些标志物可根据肿瘤样本的分子特征预测乳腺癌患者的预后。然而,数据的高维度和小样本量极大地增加了癌症生存分析的难度,这就需要开发统计方法。在这项工作中,我们提出了新颖的筛选网络方法,通过筛选关键的生存相关基因来预测患者的生存结果,并使用METABRIC数据集评估所提出方法的能力。具体而言,我们首先通过对基因表达数据使用变量筛选技术来识别基因子集。然后,我们通过纳入与所选基因子集相关的网络信息进行Cox回归分析。这项工作的新颖之处在于,在构建网络惩罚模型之前,由于采用了不同类型的筛选(即生物医学驱动、数据驱动以及两者结合),从而改进了对生存反应的预测。事实上,两种筛选方法的结合使我们能够利用现有的关于乳腺癌的生物学知识,并将其与分析所用数据中出现的额外信息相结合。此外,我们还说明了如何扩展所提出的方法以整合额外的组学层,如拷贝数变异,并且我们表明这种策略可以进一步提高我们的预测能力。总之,我们的方法能够使用少量潜在生物标志物区分高风险和低风险组的患者,因此可以帮助临床医生提供更精确的预后,并促进对有疾病风险患者的后续临床管理。