Institute of Infectious Diseases, Beijing Ditan Hospital, Capital Medical University, Beijing, China.
Beijing Key Laboratory of Emerging Infectious Diseases, Beijing, China.
PLoS Negl Trop Dis. 2018 Sep 6;12(9):e0006738. doi: 10.1371/journal.pntd.0006738. eCollection 2018 Sep.
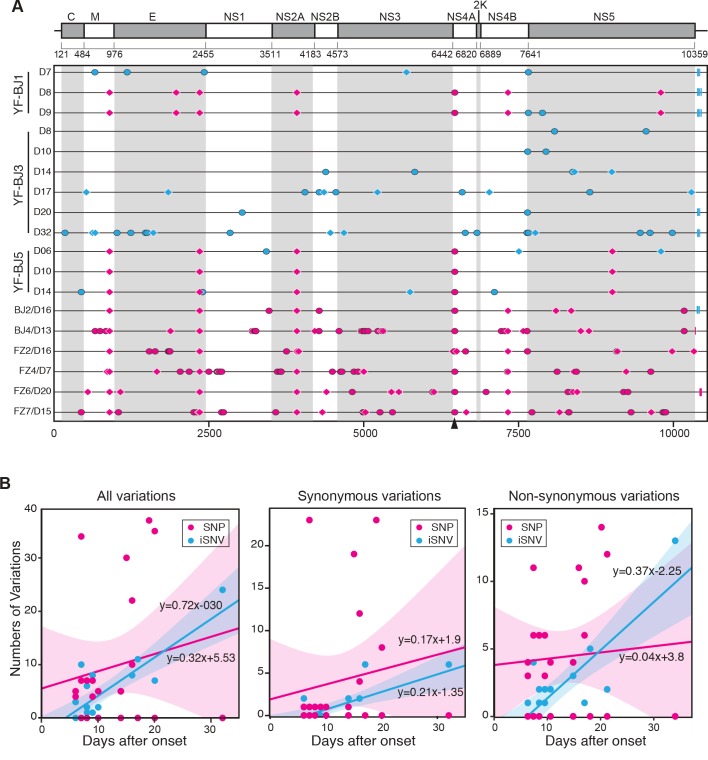
The yellow fever virus (YFV) recently reemerged in the large outbreaks in Africa and Brazil, and the first imported patients into Asia have recalled the concerns of YFV evolution. Here we show phylogenomics of YFV with serial clinical samples of the 2016 YFV infections. Phylogenetics exhibited that the 2016 strains were close to Angola 1971 strains and only three amino acid changes presented new to other lineages. Deep sequencing of viral genomes discovered 101 intrahost single nucleotide variations (iSNVs) and 234 single nucleotide polymorphisms (SNPs). Analysis of iSNV distribution and mutated allele frequency revealed that the coding regions were under purifying selection. Comparison of the evolutionary rates estimated by iSNV and SNP showed that the intrahost rate was ~2.25 times higher than the epidemic rate, and both rates were higher than the long-term YFV substitution rate, as expected. In addition, the result also hinted that short viremia duration of YFV might further hinder the evolution of YFV.
黄热病毒(YFV)近期在非洲和巴西的大规模暴发中再次出现,首例输入性亚洲患者引发了对 YFV 进化的关注。本研究对 2016 年 YFV 感染的系列临床样本进行了 YFV 的系统发育基因组学分析。系统发育分析显示,2016 年的病毒株与安哥拉 1971 年的病毒株密切相关,仅出现了 3 个氨基酸的变化,是其他谱系中没有的新变化。对病毒基因组的深度测序发现了 101 个宿主内单核苷酸变异(iSNV)和 234 个单核苷酸多态性(SNP)。iSNV 分布和突变等位基因频率的分析表明,编码区受到纯化选择。通过 iSNV 和 SNP 估计的进化率比较表明,宿主内的速率约为流行率的 2.25 倍,两者都高于长期 YFV 的替代率,这是预期的。此外,该结果还暗示,YFV 的短暂病毒血症期可能进一步阻碍 YFV 的进化。