USC Keck School of Medicine and Children's Hospital Los Angeles, 4650 Sunset Blvd, Los Angeles, CA, 90027, USA.
Children's Hospital Los Angeles, 4650 Sunset Blvd, Los Angeles, CA, 90027, USA.
Microbiome. 2018 Sep 10;6(1):155. doi: 10.1186/s40168-018-0543-z.
Recent advances in sequencing technologies and bioinformatics tools have allowed for large-scale microbiome studies that are rapidly advancing medical research. However, small changes in technique or analysis can significantly alter the results and lead to conflicting findings. Quantifying the technical versus biological variation expected in targeted 16S rRNA gene sequencing studies and how this variation changes with input biomass is critical to guide meaningful interpretation of the current literature and plan future research.
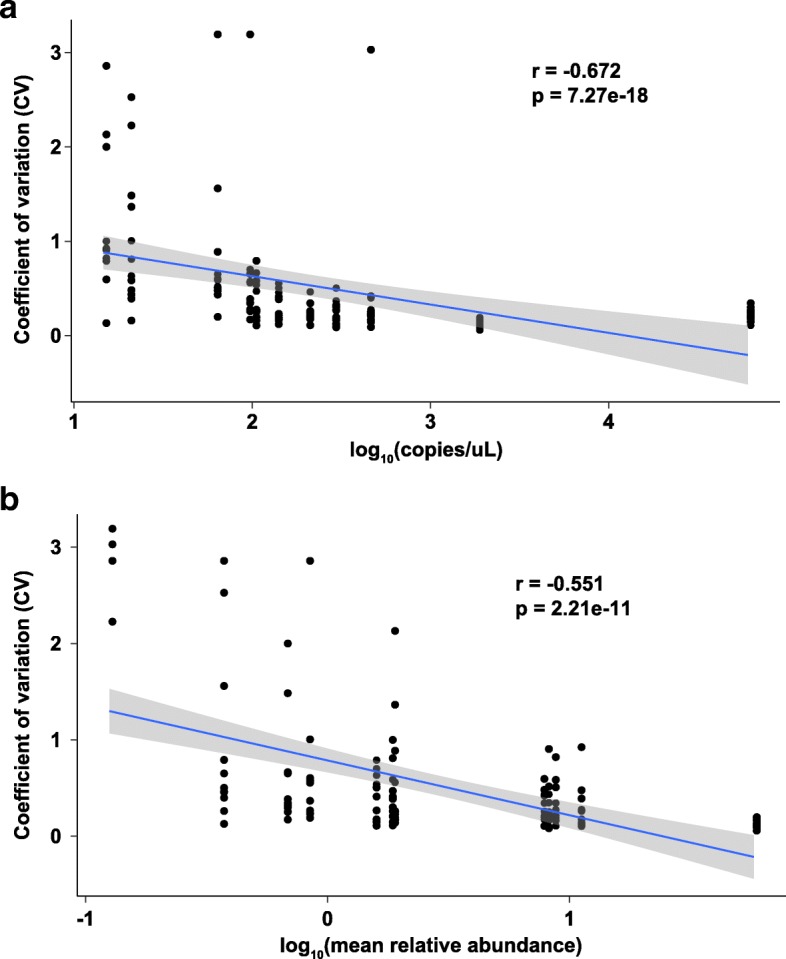
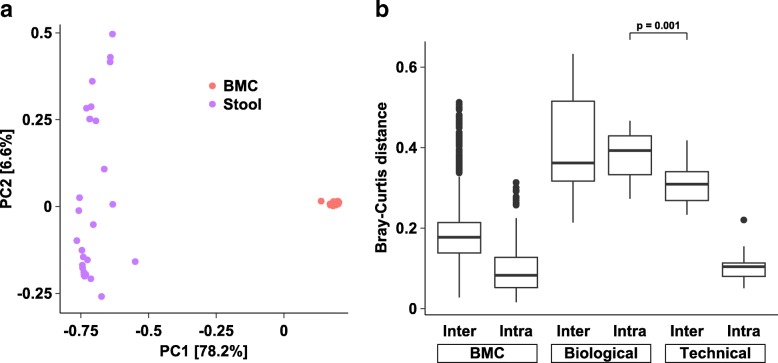
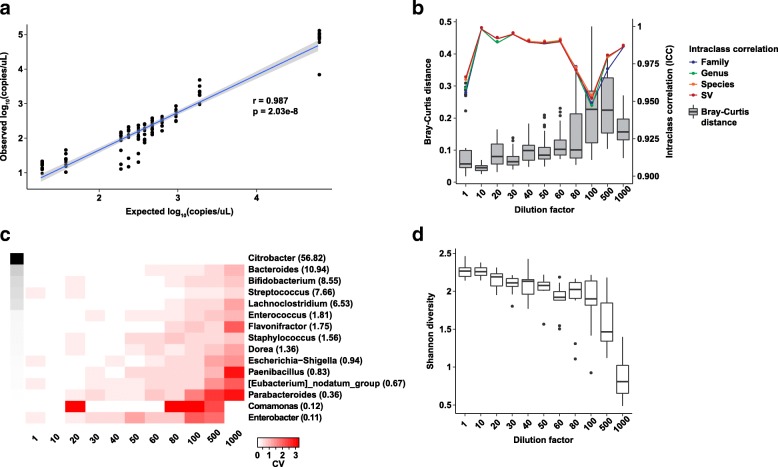
Data were compiled from 469 sequencing libraries across 19 separate targeted 16S rRNA gene sequencing runs over a 2.5-year time period. Following removal of contaminant sequences identified from negative controls, 244 samples retained sufficient reads for further analysis. Coefficients of variation for intra- and inter-assay variation from repeated measurements of a bacterial mock community ranged from 8.7 to 37.6% (intra) and 15.6 to 80.5% (inter) for all but one genus of bacteria whose relative abundance was greater than 1%. Intra- versus inter-assay Bray-Curtis pairwise distances for a single stool sample were 0.11 versus 0.31, whereas intra-assay variation from repeat stool samples from the same donor was greater at 0.38 (Wilcoxon p = 0.001). A dilution series of the bacterial mock community was used to assess the effect of input biomass on variability. Pairwise distances increased with more dilute samples, and estimates of relative abundance became unreliable below approximately 100 copies of the 16S rRNA gene per microliter. Using this data, we created a prediction model to estimate the expected variation in microbiome measurements for given input biomass and relative abundance values.
Well-controlled microbiome studies are sufficiently robust to capture small biological effects and can achieve levels of variability consistent with clinical assays. Relative abundance is negatively associated with measures of variability and has a stronger effect on variability than does absolute biomass, suggesting that it is feasible to detect differences in bacterial populations in very low-biomass samples. Further, by quantifying the effect of biomass and relative abundance on compositional variability, we developed a tool for defining the expected variance in a given microbiome study.
测序技术和生物信息学工具的最新进展使得大规模微生物组研究得以快速推进医学研究。然而,技术或分析上的微小变化会显著改变结果,并导致相互矛盾的发现。量化靶向 16S rRNA 基因测序研究中预期的技术与生物学变异性,以及这种变异性如何随输入生物量而变化,对于指导当前文献的有意义解释和规划未来的研究至关重要。
数据来自 19 个独立靶向 16S rRNA 基因测序运行的 469 个测序文库,时间跨度为 2.5 年。在去除阴性对照中鉴定出的污染序列后,有 244 个样本保留了足够的读数进行进一步分析。从细菌模拟群落的重复测量中得出的内和间测定变异系数范围为 8.7%至 37.6%(内)和 15.6%至 80.5%(间),除了一个相对丰度大于 1%的细菌属外。单个粪便样本的内-间 Bray-Curtis 成对距离为 0.11,而同一供体的重复粪便样本的内-间测定变异系数为 0.31(Wilcoxon p = 0.001)。细菌模拟群落的稀释系列用于评估输入生物量对变异性的影响。成对距离随稀释样品的增加而增加,并且在每个微升 16S rRNA 基因约 100 个拷贝以下,相对丰度的估计变得不可靠。使用这些数据,我们创建了一个预测模型,以估计给定输入生物量和相对丰度值下微生物组测量的预期变异性。
经过良好控制的微生物组研究具有足够的稳健性,可以捕获微小的生物学效应,并达到与临床检测一致的变异性水平。相对丰度与变异性测量值呈负相关,并且对变异性的影响大于绝对生物量,这表明在非常低生物量的样本中检测细菌种群的差异是可行的。此外,通过量化生物量和相对丰度对组成变异性的影响,我们开发了一种工具,用于定义给定微生物组研究中的预期方差。