Pulmonary Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20892-1590, USA.
Pulmonary Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20892-1590, USA.
Biochem Pharmacol. 2019 Sep;167:44-49. doi: 10.1016/j.bcp.2018.09.028. Epub 2018 Sep 27.
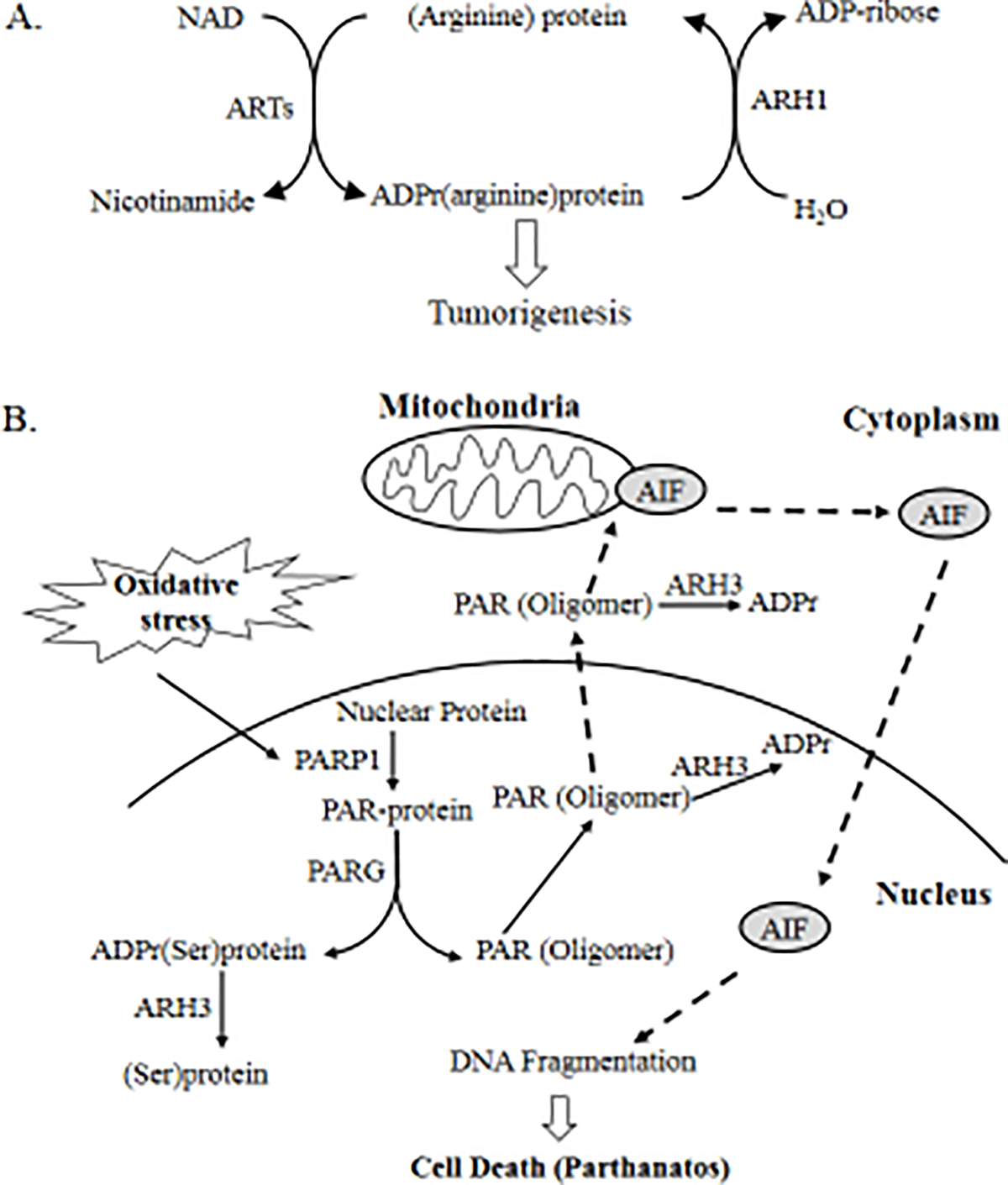
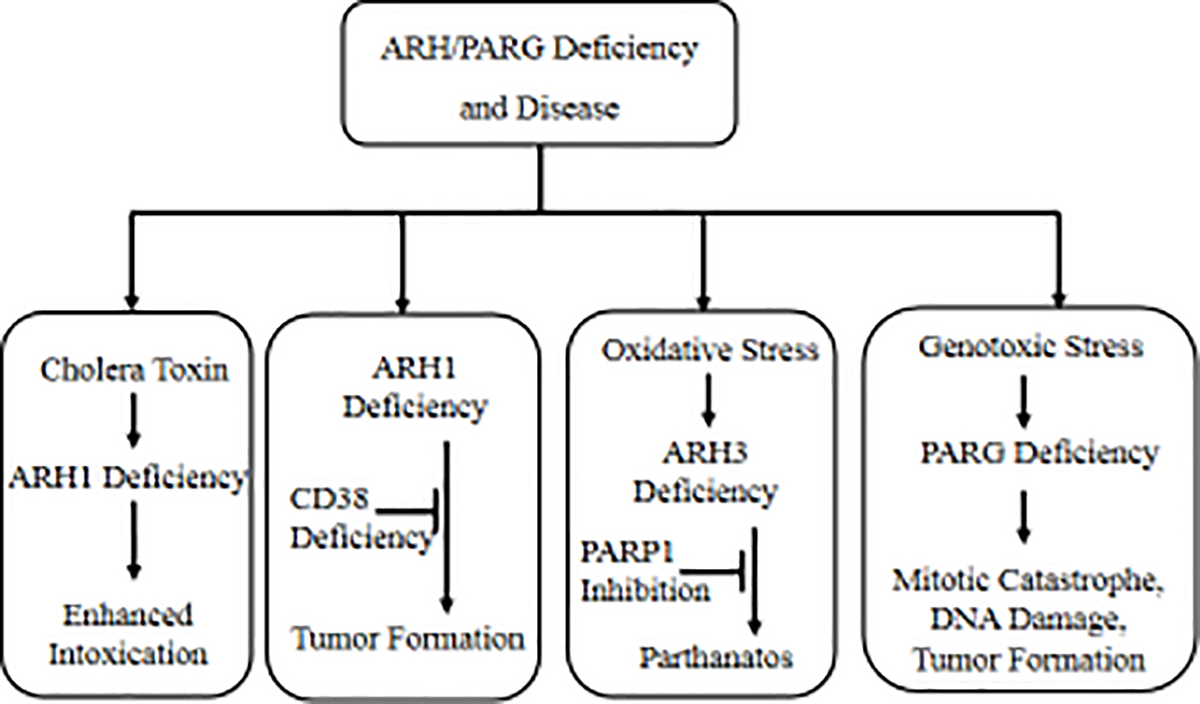
Malignant transformation may occur in the background of post-translational modification, such as ADP-ribosylation, phosphorylation and acetylation. Recent genomic analysis of ADP-ribosylation led to the discovery of more than twenty ADP-ribosyltransferases (ARTs), which catalyze either mono- or poly-ADP-ribosylation. ARTs catalyze the attachment of ADP-ribose to acceptor molecules. The ADP-ribose-acceptor bond can then be cleaved by a family of hydrolases in a substrate-specific manner, which is dependent on the acceptor and its functional group, e.g., arginine (guanidino), serine (hydroxyl), aspartate (carboxyl). These hydrolases vary in structure and function, and include poly-ADP-ribose glycohydrolase (PARG), MacroD1, MacroD2, terminal ADP-ribose protein glycohydrolase 1 (TARG1) and ADP-ribosyl-acceptor hydrolases (ARHs). In murine models, PARG deficiency increased susceptibility to alkylating agents-induced carcinogenesis. Similarly, by cleaving mono-ADP-ribosylated arginine on target proteins, ARH1 appears to inhibit tumor formation, suggesting that ARH1 is a tumor-suppressor gene. Although ARH3 is similar to ARH1 in amino acid sequence and crystal structure, ARH3 does not cleave ADP-ribose-arginine, rather it degrades in an exocidic manner, the PAR polymer and cleaves O-acetyl-ADP-ribose (OAADPr) and the ADP-ribose-serine linkage in acceptor proteins. Under conditions of oxidative stress, ARH3-deficient cells showed increased cytosolic PAR accumulation and PARP-1 mediated cell death. These findings expand our understanding of ADP-ribosylation and provide new therapeutic targets for cancer treatment. In the present review, research on ARH1-regulated tumorigenesis and cell death pathways that are enhanced by ARH3 deficiency are discussed.
恶性转化可能发生在翻译后修饰的背景下,如 ADP-核糖基化、磷酸化和乙酰化。最近对 ADP-核糖基化的基因组分析导致发现了二十多种 ADP-核糖基转移酶(ARTs),它们催化单或多 ADP-核糖基化。ARTs 催化 ADP-核糖基与受体分子的连接。然后,ADP-核糖-受体键可以通过一系列以底物特异性方式切割的水解酶,这取决于受体及其功能基团,例如精氨酸(胍基)、丝氨酸(羟基)、天冬氨酸(羧基)。这些水解酶在结构和功能上有所不同,包括多 ADP-核糖基糖水解酶(PARG)、MacroD1、MacroD2、末端 ADP-核糖基蛋白糖水解酶 1(TARG1)和 ADP-核糖基受体水解酶(ARHs)。在小鼠模型中,PARG 缺乏会增加对烷化剂诱导的致癌作用的易感性。同样,通过切割靶蛋白上的单 ADP-核糖基精氨酸,ARH1 似乎抑制肿瘤形成,表明 ARH1 是一种肿瘤抑制基因。尽管 ARH3 在氨基酸序列和晶体结构上与 ARH1 相似,但 ARH3 不会切割 ADP-核糖基-精氨酸,而是以 exocidic 方式降解,PAR 聚合物并切割 O-乙酰-ADP-核糖基(OAADPr)和受体蛋白中的 ADP-核糖基-丝氨酸键。在氧化应激条件下,ARH3 缺乏的细胞显示细胞浆 PAR 积累增加和 PARP-1 介导的细胞死亡。这些发现扩展了我们对 ADP-核糖基化的理解,并为癌症治疗提供了新的治疗靶点。在本综述中,讨论了 ARH3 缺乏增强的 ARH1 调节的肿瘤发生和细胞死亡途径的研究。