Nagpal Sunil, Haque Mohammed Monzoorul, Singh Rashmi, Mande Sharmila S
Bio-Sciences R&D Division, TCS Research, Tata Consultancy Services, Pune, India.
Front Microbiol. 2019 Jan 14;9:3336. doi: 10.3389/fmicb.2018.03336. eCollection 2018.
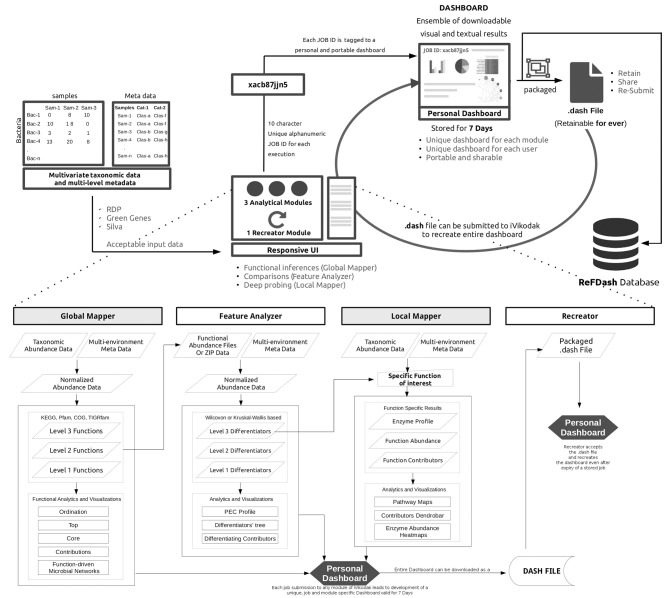
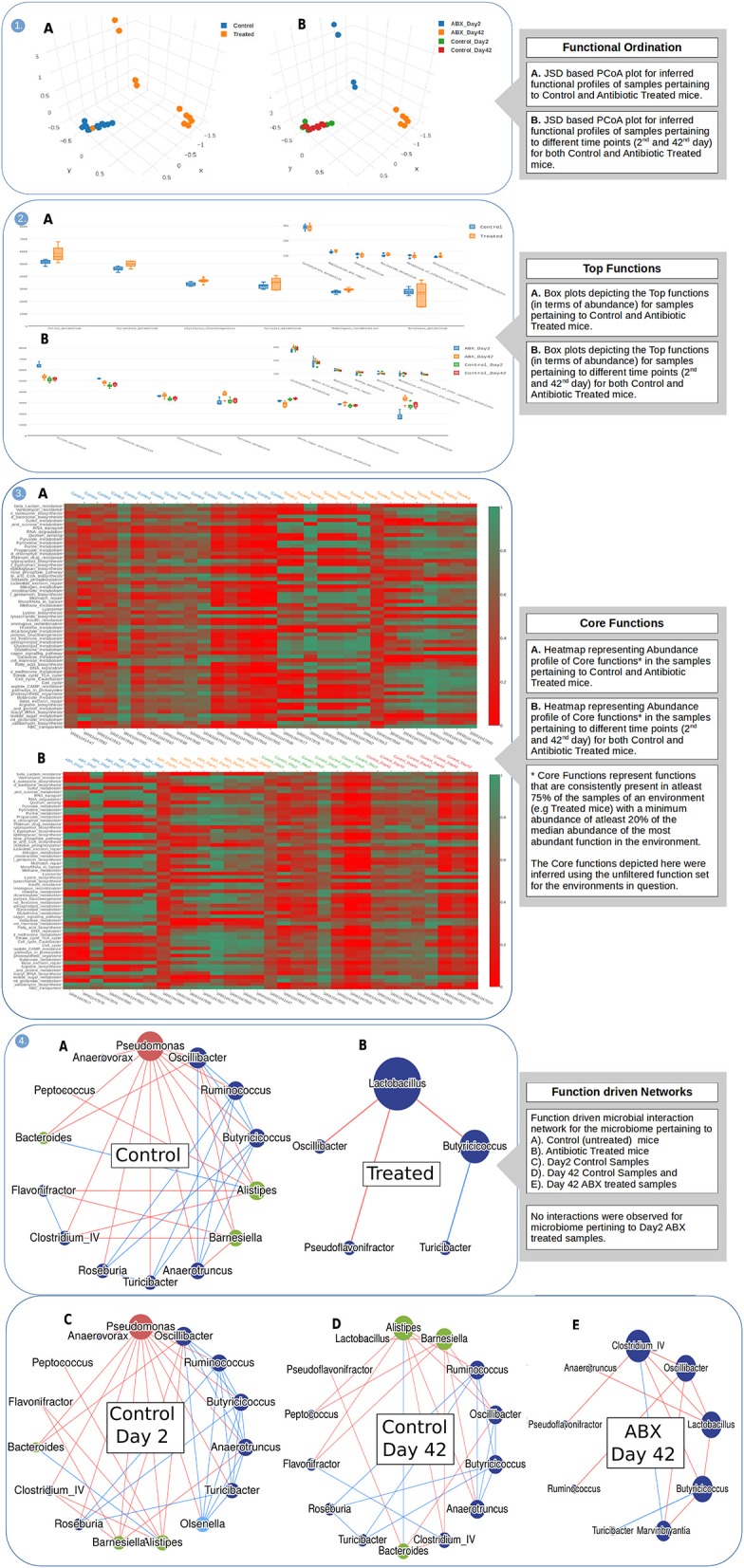
The objectives of any metagenomic study typically include identification of resident microbes and their relative proportions (taxonomic analysis), profiling functional diversity (functional analysis), and comparing the identified microbes and functions with available metadata (comparative metagenomics). Given the advantage of cost-effectiveness and convenient data-size, amplicon-based sequencing has remained the technology of choice for exploring phylogenetic diversity of an environment. A recent school of thought, employing the existing genome annotation information for inferring functional capacity of an identified microbiome community, has given a promising alternative to Whole Genome Shotgun sequencing for functional analysis. Although a handful of tools are currently available for function inference, their scope, functionality and utility has essentially remained limited. Need for a comprehensive framework that expands upon the existing scope and enables a standardized workflow for function inference, analysis, and visualization, is therefore felt. We present iVikodak, a multi-modular web-platform that hosts a logically inter-connected repertoire of functional inference and analysis tools, coupled with a comprehensive visualization interface. iVikodak is equipped with microbial co-inhabitance pattern driven published algorithms along with multiple updated databases of various curated microbe-function maps. It also features an advanced task management and result sharing system through introduction of personalized and portable dashboards. In addition to inferring functions from 16S rRNA gene data, iVikodak enables (a) an in-depth analysis of specific functions of interest (b) identification of microbes contributing to various functions (c) microbial interaction patterns through function-driven correlation networks, and (d) simultaneous functional comparison between multiple microbial communities. We have bench-marked iVikodak through multiple case studies and comparisons with existing state of art. We also introduce the concept of a public repository which provides a first of its kind community-driven framework for scientific data analytics, collaboration and sharing in this area of microbiome research. Developed using modern design and task management practices, iVikodak provides a multi-modular, yet inter-operable, one-stop framework, that intends to simplify the entire approach toward inferred function analysis. It is anticipated to serve as a significant value addition to the existing space of functional metagenomics. iVikodak web-server may be freely accessed at https://web.rniapps.net/iVikodak/.
任何宏基因组学研究的目标通常包括识别常驻微生物及其相对比例(分类分析)、描绘功能多样性(功能分析),以及将识别出的微生物和功能与可用的元数据进行比较(比较宏基因组学)。鉴于基于扩增子测序具有成本效益高和数据量便捷的优势,它一直是探索环境系统发育多样性的首选技术。最近有一种思路,利用现有的基因组注释信息来推断已识别微生物群落的功能能力,这为全基因组鸟枪法测序进行功能分析提供了一个有前景的替代方案。尽管目前有一些工具可用于功能推断,但其范围、功能和实用性基本上仍然有限。因此,需要一个全面框架,在现有范围的基础上进行扩展,并为功能推断、分析和可视化提供标准化工作流程。我们展示了iVikodak,这是一个多模块网络平台,拥有一系列逻辑上相互连接的功能推断和分析工具,以及一个全面的可视化界面。iVikodak配备了由微生物共生模式驱动的已发表算法,以及多个更新的各种精心策划的微生物 - 功能图谱数据库。它还通过引入个性化和便携式仪表板,具有先进的任务管理和结果共享系统。除了从16S rRNA基因数据推断功能外,iVikodak还能够(a)对感兴趣的特定功能进行深入分析,(b)识别对各种功能有贡献的微生物,(c)通过功能驱动的相关网络分析微生物相互作用模式,以及(d)同时对多个微生物群落进行功能比较。我们通过多个案例研究以及与现有先进技术的比较,对iVikodak进行了基准测试。我们还引入了公共存储库的概念,它为微生物组研究领域的科学数据分析、协作和共享提供了首个由社区驱动的框架。iVikodak采用现代设计和任务管理实践开发,提供了一个多模块但可互操作的一站式框架,旨在简化整个功能推断分析方法。预计它将为现有的功能宏基因组学领域带来显著的价值提升。可通过https://web.rniapps.net/iVikodak/免费访问iVikodak网络服务器。