Department of Internal Medicine, Radboud University Medical Center, Nijmegen, the Netherlands.
Radboud Institute for Molecular Life Sciences (RIMLS), Radboud University Medical Center, Nijmegen, the Netherlands.
Arthritis Res Ther. 2019 Feb 6;21(1):50. doi: 10.1186/s13075-019-1834-x.
Acute gouty arthritis currently is the most common form of inflammatory arthritis in developed countries. Treatment is still suboptimal. Dosage of urate-lowering therapy is often too low to reach target urate levels, and adherence to therapy is poor. In this study, we therefore explore a new treatment option to limit inflammation in acute gout: specific histone deacetylase (HDAC) inhibition.
Peripheral blood mononuclear cells (PBMCs) were cultured with a combination of monosodium urate crystals (MSU) and palmitic acid (C16.0) in order to activate the NLRP3 inflammasome and induce IL-1β production. HDAC inhibitors and other compounds were added beforehand with a 1-h pre-incubation period.
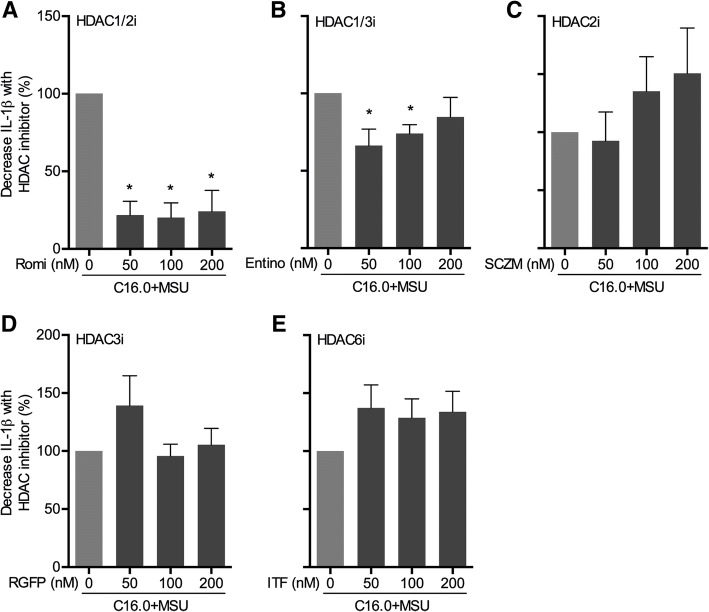
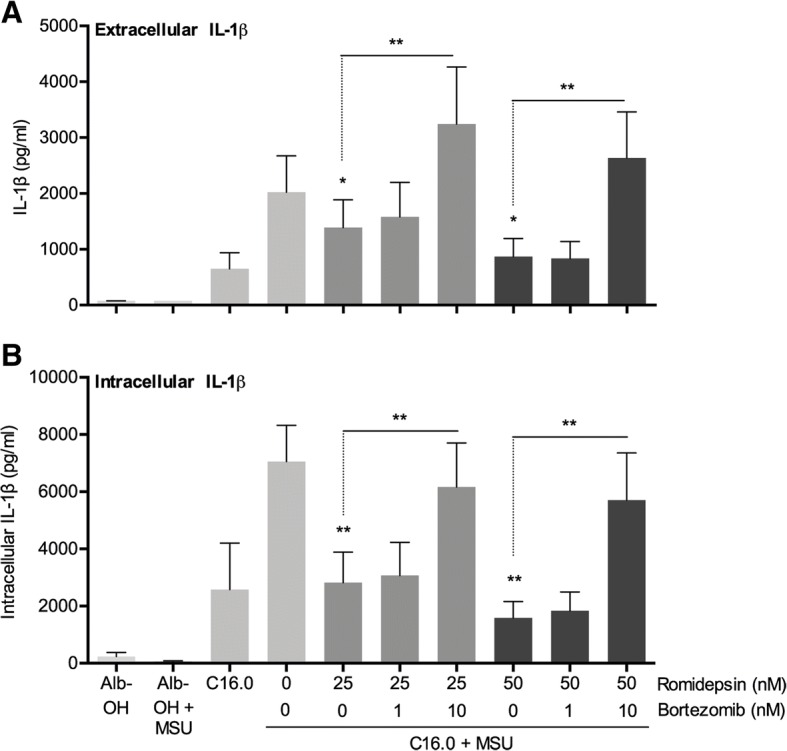
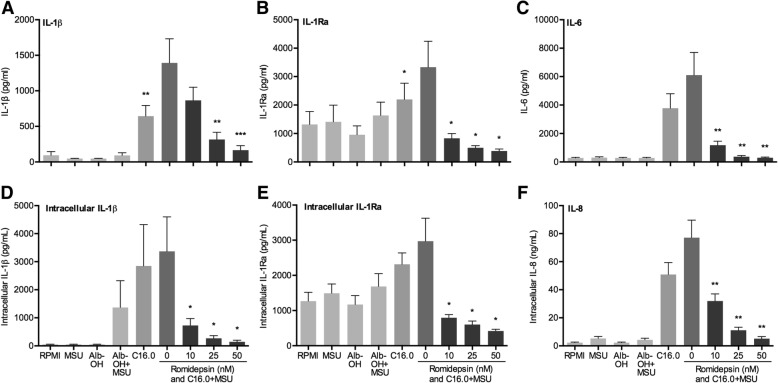
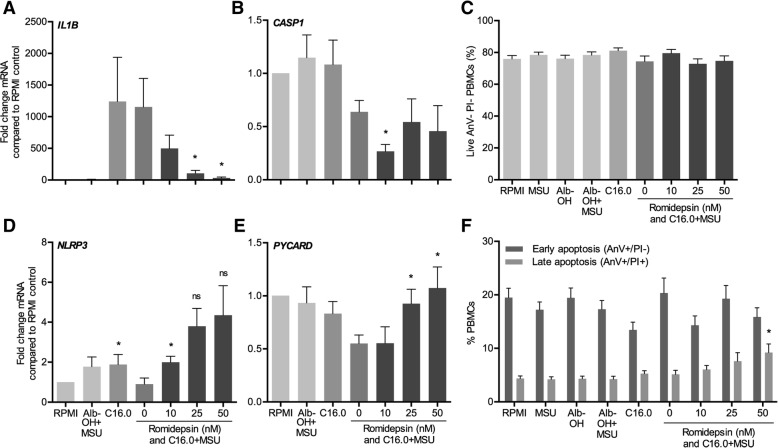
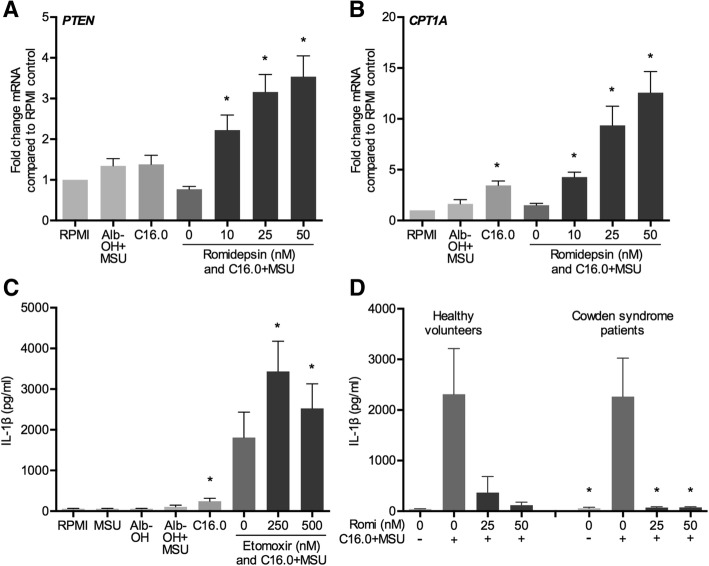
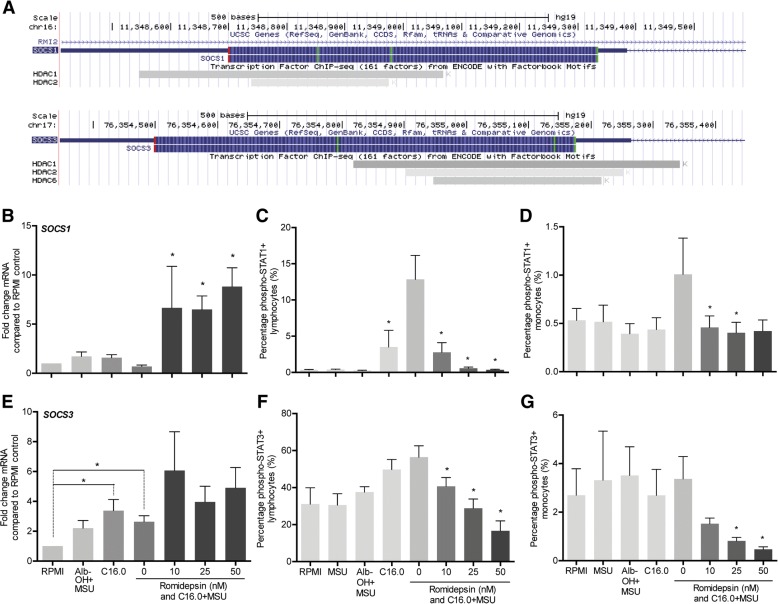
The HDAC1/2 inhibitor romidepsin was most potent in lowering C16.0+MSU-induced IL-1β production compared to other specific class I HDAC inhibitors. At 10 nM, romidepsin decreased IL-1β, IL-1Ra, IL-6, and IL-8 production. IL-1β mRNA was significantly decreased at 25 nM. Although romidepsin increased PTEN expression, PBMCs from patients with germline mutations in PTEN still responded well to romidepsin. Romidepsin also increased SOCS1 expression and blocked STAT1 and STAT3 activation. Furthermore, experiments with bortezomib showed that blocking the proteasome reverses the cytokine suppression by romidepsin.
Our results show that romidepsin is a very potent inhibitor of C16.0+MSU-induced cytokines in vitro. Romidepsin upregulated transcription of SOCS1, which was shown to directly target inflammatory signaling molecules for proteasomal degradation. Inhibiting the proteasome therefore reversed the cytokine-suppressive effects of romidepsin. HDAC1/2 dual inhibition could therefore be a highly potent new treatment option for acute gout, although safety has to be determined in vivo.
急性痛风性关节炎目前是发达国家最常见的炎症性关节炎形式。治疗仍不尽如人意。降尿酸治疗的剂量往往太低,无法达到目标尿酸水平,而且治疗的依从性也很差。因此,在这项研究中,我们探索了一种新的治疗方法来限制急性痛风的炎症:特定组蛋白去乙酰化酶(HDAC)抑制。
外周血单核细胞(PBMCs)与单钠尿酸盐晶体(MSU)和棕榈酸(C16.0)联合培养,以激活 NLRP3 炎性体并诱导 IL-1β 产生。HDAC 抑制剂和其他化合物在孵育前加入 1 小时。
HDAC1/2 抑制剂罗米地辛在降低 C16.0+MSU 诱导的 IL-1β 产生方面比其他特异性 I 类 HDAC 抑制剂更有效。在 10 nM 时,罗米地辛降低了 IL-1β、IL-1Ra、IL-6 和 IL-8 的产生。25 nM 时,IL-1βmRNA 显著降低。尽管罗米地辛增加了 PTEN 的表达,但来自 PTEN 种系突变患者的 PBMCs 仍然对罗米地辛反应良好。罗米地辛还增加了 SOCS1 的表达,并阻断了 STAT1 和 STAT3 的激活。此外,用硼替佐米进行的实验表明,阻断蛋白酶体可逆转罗米地辛对细胞因子的抑制作用。
我们的结果表明,罗米地辛是体外 C16.0+MSU 诱导细胞因子的非常有效的抑制剂。罗米地辛上调了 SOCS1 的转录,SOCS1 被证明可直接针对炎性信号分子进行蛋白酶体降解。因此,抑制蛋白酶体可逆转罗米地辛的细胞因子抑制作用。因此,HDAC1/2 双重抑制可能是急性痛风的一种非常有效的新治疗方法,尽管还需要在体内确定安全性。