Department of Automation, Harbin Engineering University, Harbin, China.
Department of Neurosurgery, Stanford University, California, USA.
Biol Direct. 2019 Feb 13;14(1):4. doi: 10.1186/s13062-018-0229-2.
More than 90% of neuroblastoma patients are cured in the low-risk group while only less than 50% for those with high-risk disease can be cured. Since the high-risk patients still have poor outcomes, we need more accurate stratification to establish an individualized precise treatment plan for the patients to improve the long-term survival rate.
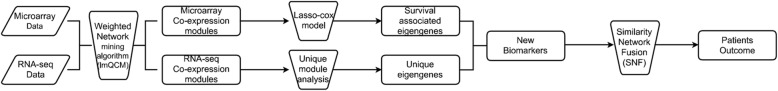
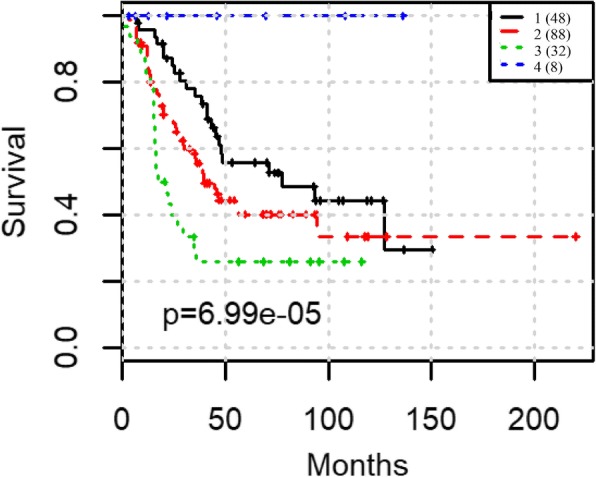
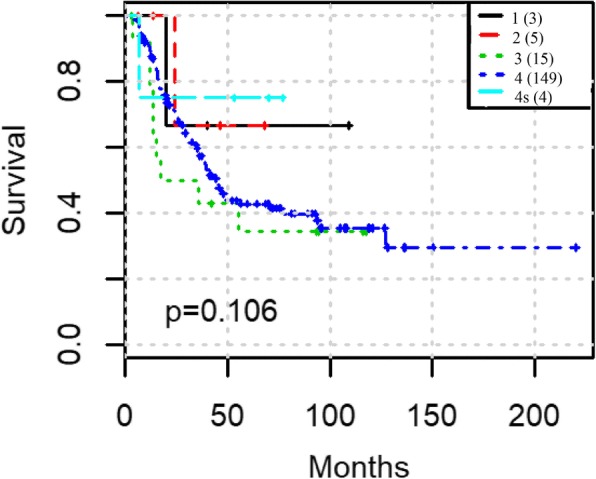
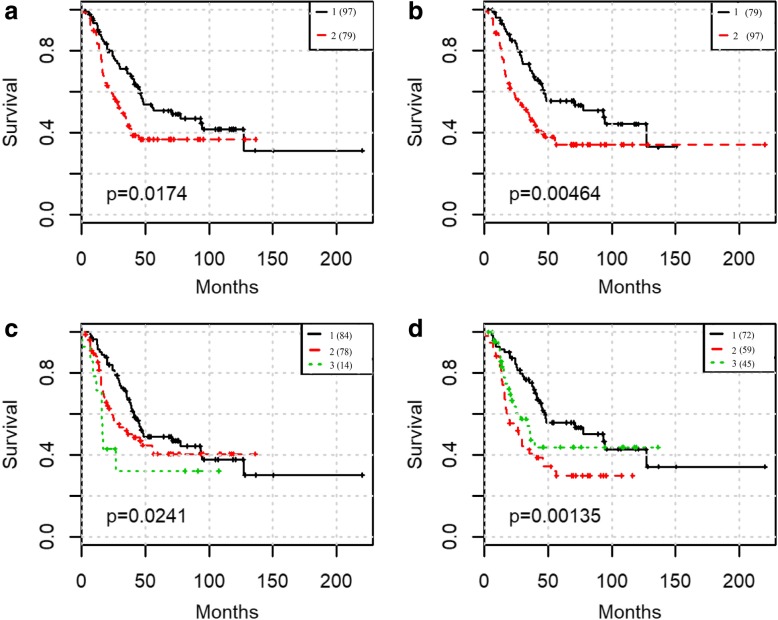
We focus on extracting features and providing a workflow to improve survival prediction for neuroblastoma patients. With a workflow for gene co-expression network (GCN) mining in microarray and RNA-Seq datasets, we extracted molecular features from each co-expressed module and summarized them into eigengenes. Then we adopted the lasso-regularized Cox proportional hazards model to select the most informative eigengene features regarding association to the risk of metastasis. Nine eigengenes were selected which show strong association with patient survival prognosis. All of the nine corresponding gene modules also have highly enriched biological functions or cytoband locations. Three of them are unique modules to RNA-Seq data, which complement the modules from microarray data in terms of survival prognosis. We then merged all eigengenes from these unique modules and used an integrative method called Similarity Network Fusion to test the prognostic power of these eigengenes for prognosis. The prognostic accuracies are significantly improved as compared to using all eigengenes, and a subgroup of patients with very poor survival rate was identified.
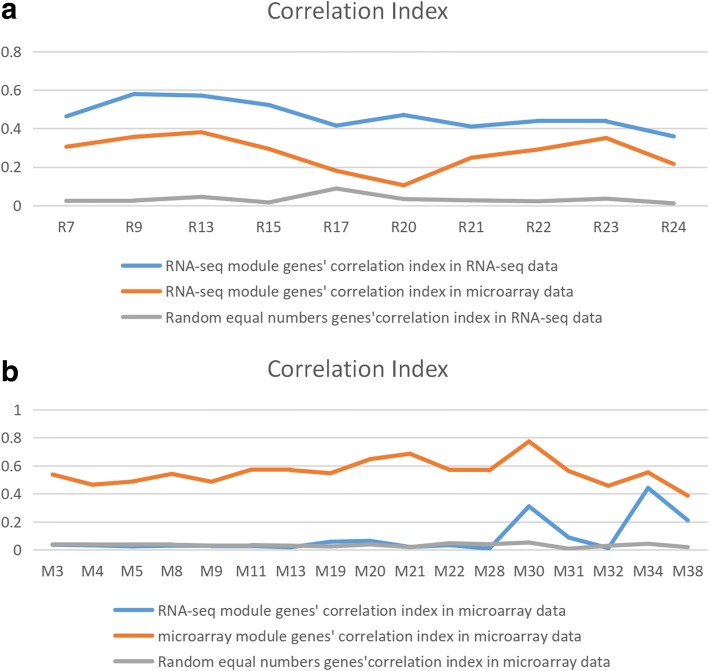
We first compared GCNs mined from microarray and RNA-seq data. We discovered that each data modality yields unique GCNs, which are enriched with clear biological functions. Then we do module unique analysis and use lasso-cox model to select survival-associated eigengenes. Integration of unique and survival-associated eigengenes from both data types provides complementary information that leads to more accurate survival prognosis.
Reviewed by Susmita Datta, Marco Chierici and Dimitar Vassilev.
超过 90%的神经母细胞瘤患者在低危组中得到治愈,而高危疾病患者中只有不到 50%能够治愈。由于高危患者的预后仍然较差,我们需要更准确的分层,为患者制定个体化的精确治疗计划,以提高长期生存率。
我们专注于提取特征并提供工作流程,以提高神经母细胞瘤患者的生存预测。通过微阵列和 RNA-Seq 数据集的基因共表达网络(GCN)挖掘工作流程,我们从每个共表达模块中提取分子特征,并将其总结为特征基因。然后,我们采用套索正则化 Cox 比例风险模型,选择与转移风险相关的最具信息量的特征基因特征。选择了九个特征基因,它们与患者生存预后有很强的关联。所有九个对应的基因模块也具有高度丰富的生物学功能或细胞带位置。其中三个是 RNA-Seq 数据特有的模块,它们在生存预后方面补充了微阵列数据的模块。然后,我们合并了这些独特模块的所有特征基因,并使用一种名为相似性网络融合的综合方法来测试这些特征基因对预后的预测能力。与使用所有特征基因相比,预后准确性得到了显著提高,并确定了一组生存预后非常差的患者亚组。
我们首先比较了从微阵列和 RNA-seq 数据中挖掘出的 GCN。我们发现每种数据模式都产生独特的 GCN,这些 GCN 富含明确的生物学功能。然后,我们进行模块特有分析,并使用套索 Cox 模型选择与生存相关的特征基因。来自两种数据类型的独特和与生存相关的特征基因的整合提供了互补信息,从而导致更准确的生存预测。
由 Susmita Datta、Marco Chierici 和 Dimitar Vassilev 评审。