Quan Jianping, Cai Gengyuan, Yang Ming, Zeng Zhonghua, Ding Rongrong, Wang Xingwang, Zhuang Zhanwei, Zhou Shenping, Li Shaoyun, Yang Huaqiang, Li Zicong, Zheng Enqin, Huang Wen, Yang Jie, Wu Zhenfang
College of Animal Science and National Engineering Research Center for Breeding Swine Industry, South China Agricultural University, Guangzhou, China.
National Engineering Research Center for Breeding Swine Industry, Guangdong Wens Foodstuffs Group Co., Ltd., Guangzhou, China.
Front Microbiol. 2019 Jan 29;10:52. doi: 10.3389/fmicb.2019.00052. eCollection 2019.
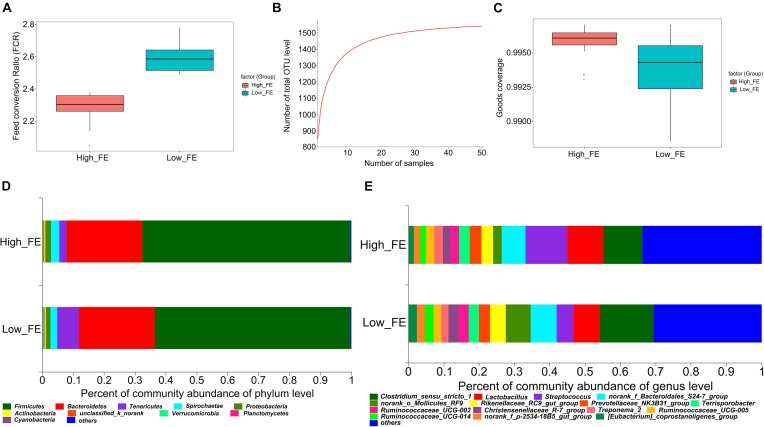
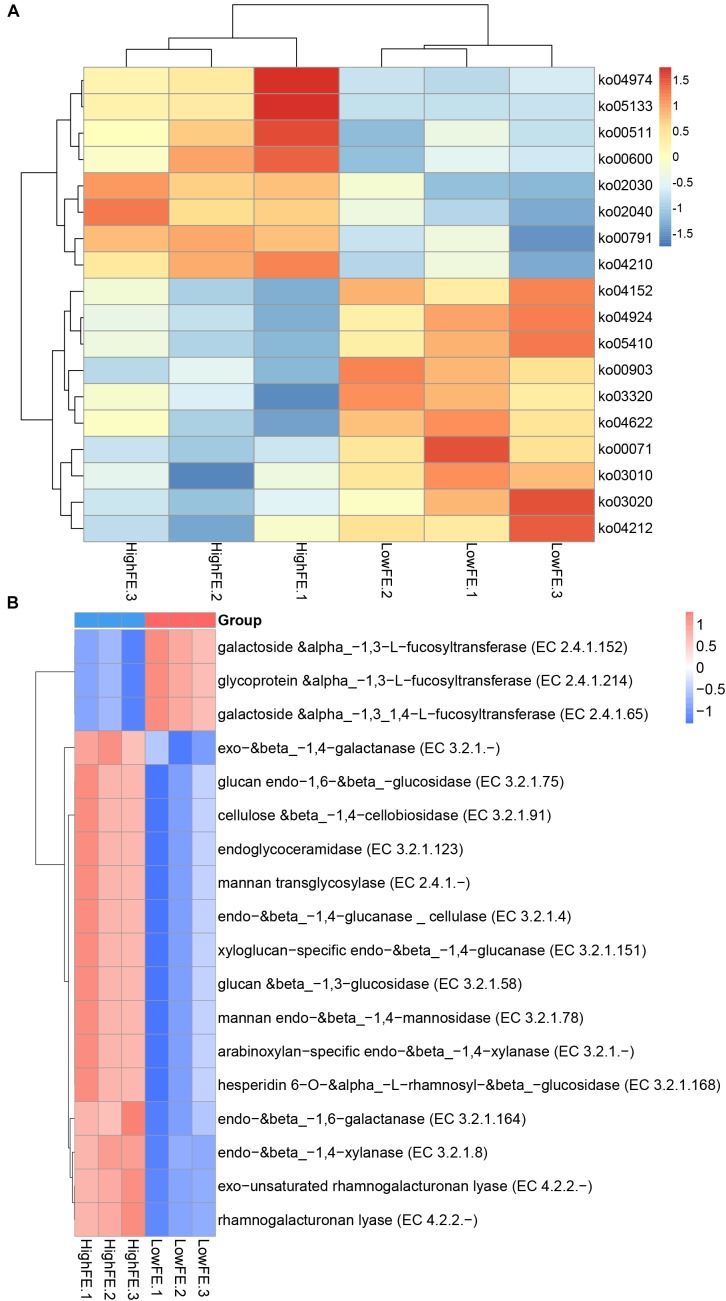
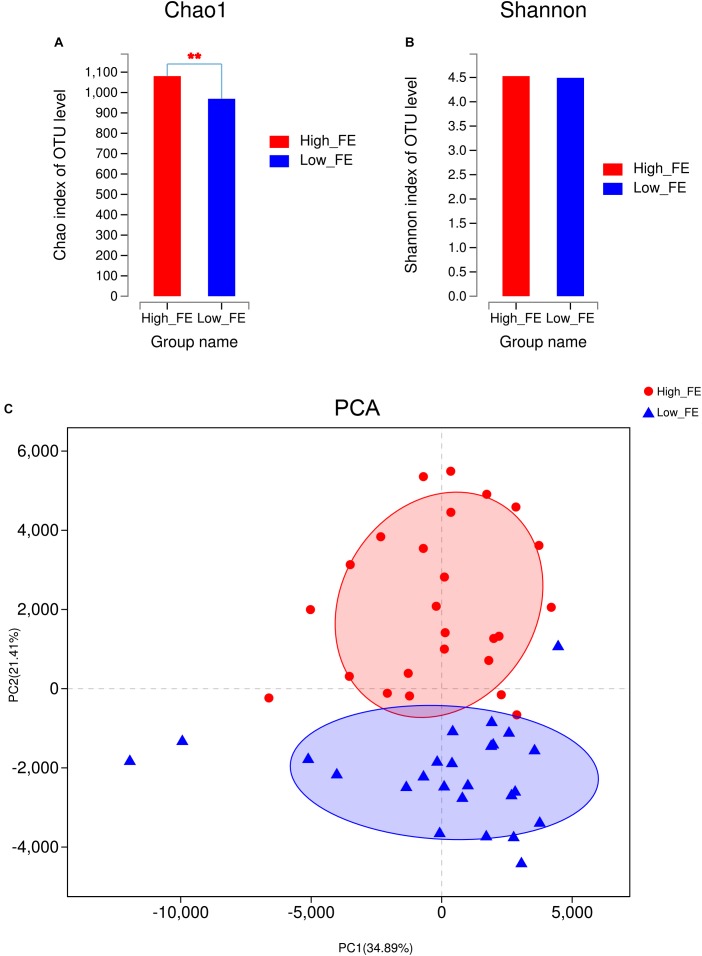
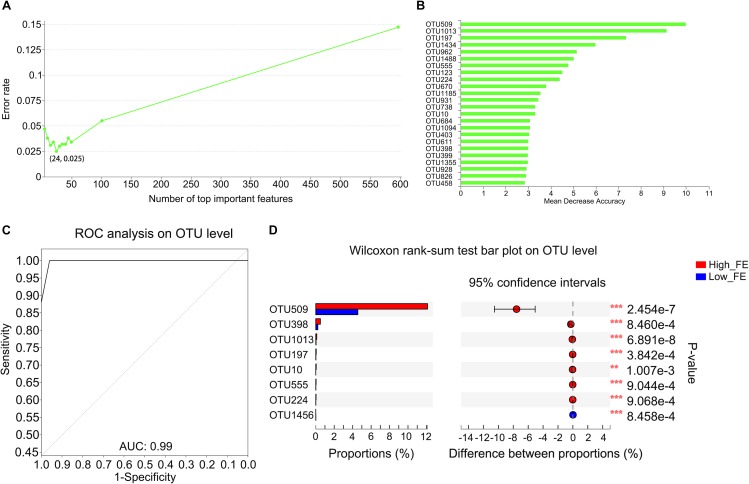
Gut microbiota has indispensable roles in nutrient digestion and energy harvesting, especially in processing the indigestible components of dietary polysaccharides. Searching for the microbial taxa and functional capacity of the gut microbiome associated with feed efficiency (FE) can provide important knowledge to increase profitability and sustainability of the swine industry. In the current study, we performed a comparative analysis of the fecal microbiota in 50 commercial Duroc × (Landrace × Yorkshire) (DLY) pigs with polarizing FE using 16S rRNA gene sequencing and shotgun metagenomic sequencing. There was a different microbial community structure in the fecal microbiota of pigs with different FE. Random forest analysis identified 24 operational taxonomic units (OTUs) as potential biomarkers to improve swine FE. Multiple comparison analysis detected 8 OTUs with a significant difference or tendency toward a difference between high- and low-FE pigs ( < 0.01, < 0.1). The high-FE pigs had a greater abundance of OTUs that were from the Lachnospiraceae and Prevotellaceae families and the - and genera than low-FE pigs. A sub-species subsp. could be an important candidate for improving FE. The functional capacity analysis found 18 KEGG pathways and CAZy EC activities that were different between high- and low-FE pigs. The fecal microbiota in high FE pigs have greater functional capacity to degrade dietary cellulose, polysaccharides, and protein and may have a greater abundance of microbes that can promote intestinal health. These results provided insights for improving porcine FE through modulating the gut microbiome.
肠道微生物群在营养消化和能量获取中发挥着不可或缺的作用,尤其是在处理膳食多糖的不可消化成分方面。寻找与饲料效率(FE)相关的肠道微生物群的微生物分类群和功能能力,可以为提高养猪业的盈利能力和可持续性提供重要知识。在本研究中,我们使用16S rRNA基因测序和鸟枪法宏基因组测序,对50头具有两极分化FE的商业杜洛克×(长白×约克夏)(DLY)猪的粪便微生物群进行了比较分析。不同FE的猪的粪便微生物群中存在不同的微生物群落结构。随机森林分析确定了24个可操作分类单元(OTUs)作为改善猪FE的潜在生物标志物。多重比较分析检测到8个OTUs在高FE和低FE猪之间存在显著差异或差异趋势(<0.01,<0.1)。高FE猪比低FE猪具有更多来自毛螺菌科和普雷沃氏菌科以及-和-属的OTUs。一个亚种亚种可能是改善FE的重要候选者。功能能力分析发现高FE和低FE猪之间有18条KEGG途径和CAZy EC活性不同。高FE猪的粪便微生物群具有更强的降解膳食纤维素、多糖和蛋白质的功能能力,并且可能有更多能促进肠道健康的微生物。这些结果为通过调节肠道微生物群改善猪的FE提供了见解。