Huang Anni, Cai Rujian, Wang Qun, Shi Lei, Li Chunling, Yan He
School of Food Science and Engineering, South China University of Technology, Guangzhou, China.
Institute of Animal Health, Guangdong Academy of Agricultural Sciences, Guangzhou, China.
Front Microbiol. 2019 Feb 25;10:322. doi: 10.3389/fmicb.2019.00322. eCollection 2019.
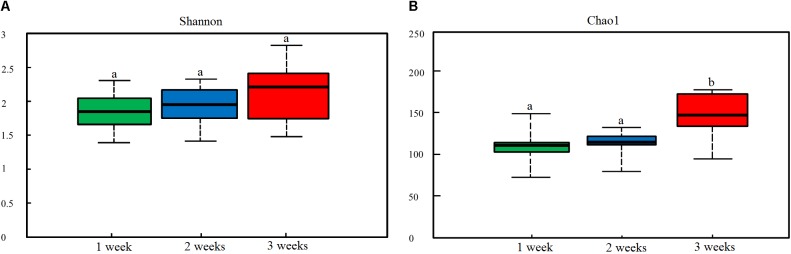
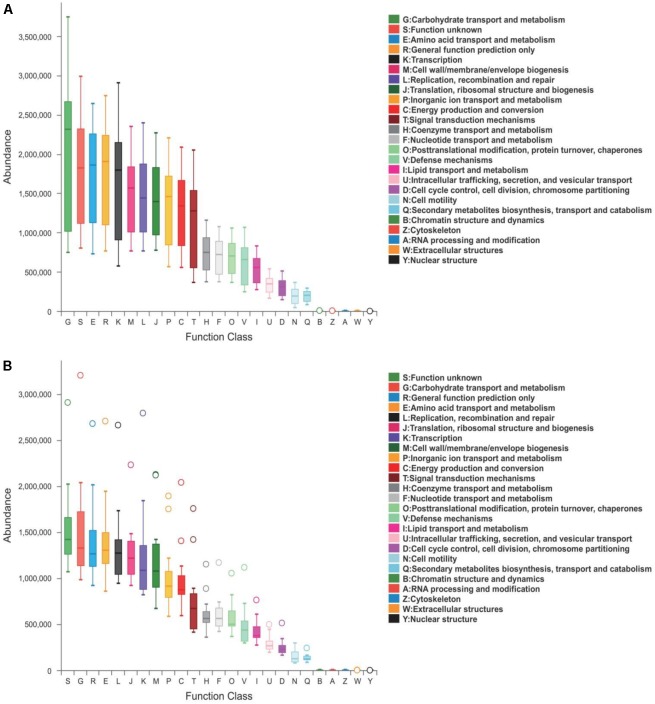
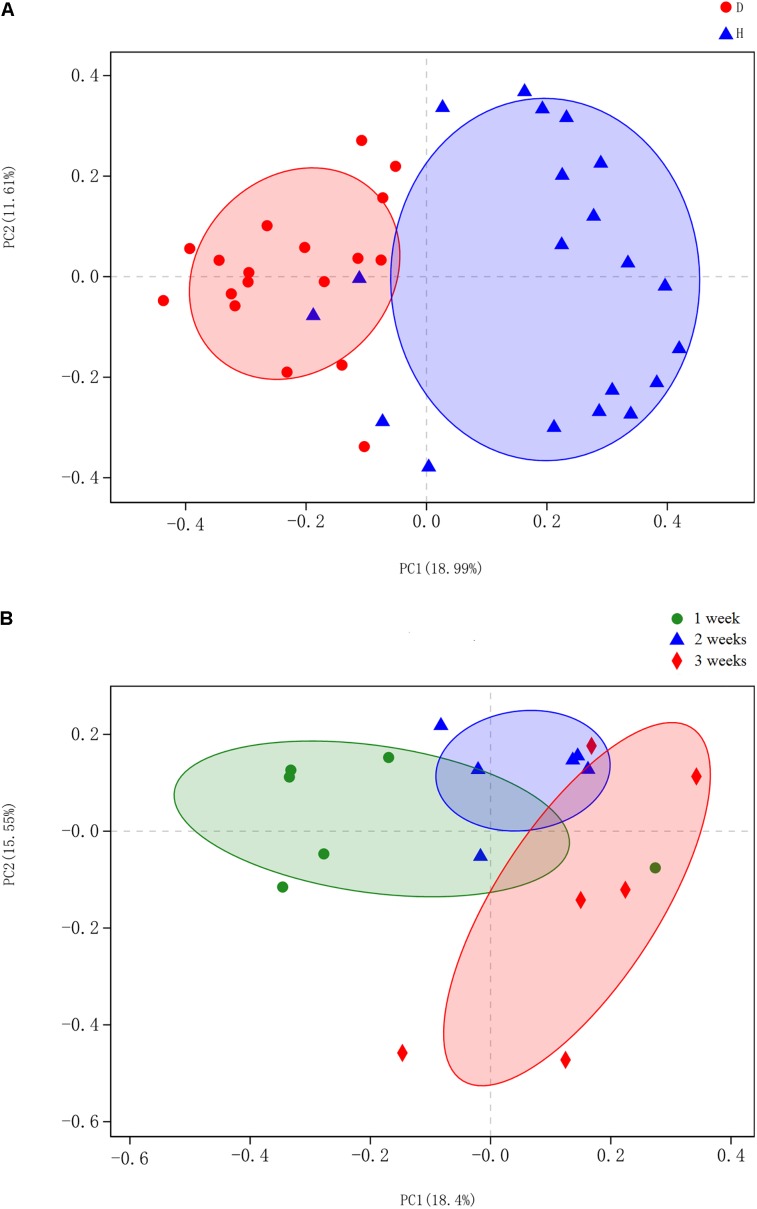
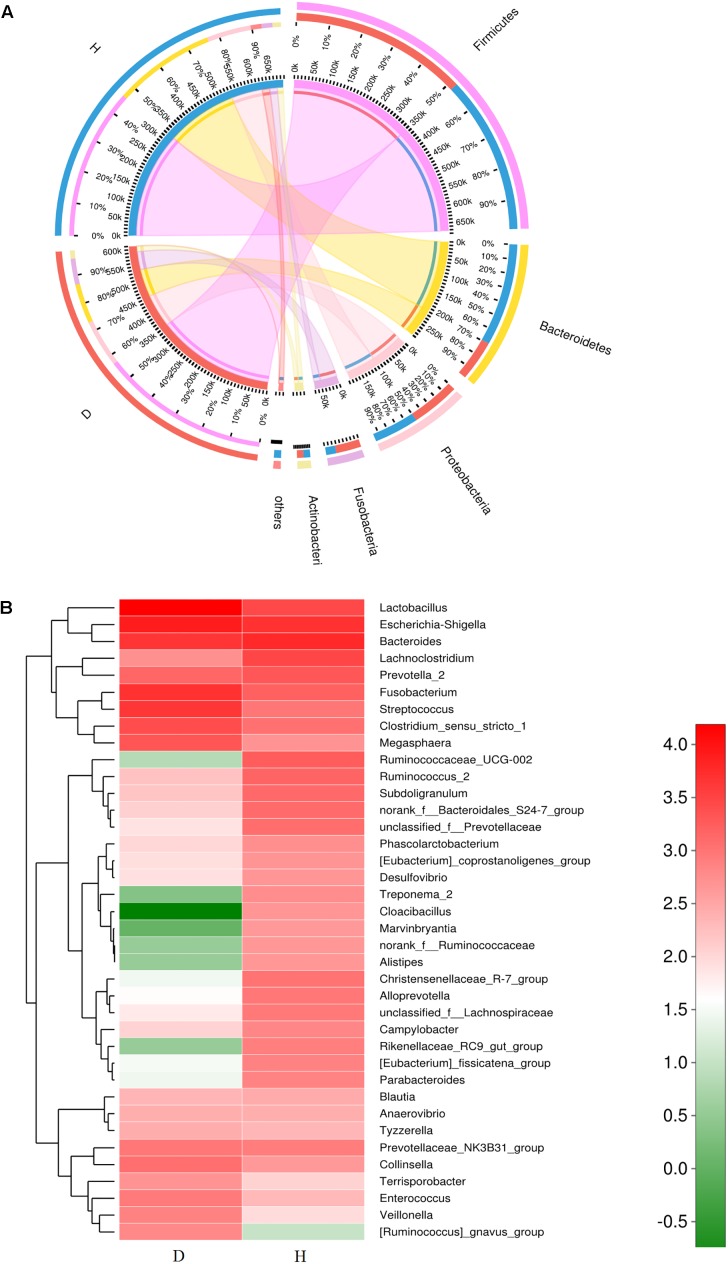
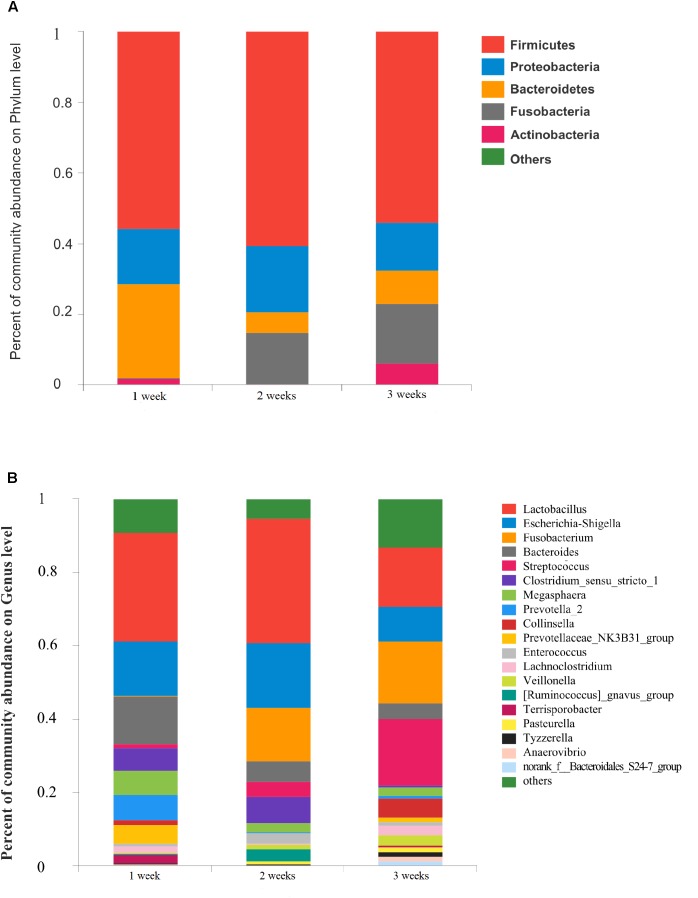
Porcine epidemic diarrhea (PED) is a disease that has a devastating effect on livestock. Currently, most studies are focused on comparing gut microbiota of healthy piglets and piglets with PED, resulting in gut microbial populations related to dynamic change in diarrheal piglets being poorly understood. The current study analyzed the characteristics of gut microbiota in porcine epidemic diarrhea virus (PEDV)-infected piglets during the suckling transition stage. Fresh fecal samples were collected from 1 to 3-week-old healthy piglets ( = 20) and PEDV infected piglets ( = 18) from the same swine farm. Total DNA was extracted from each sample and the V3-V4 hypervariable region of the 16S rRNA gene was amplified and sequenced using the Illumina MiSeq platform. Statistically significant differences were observed in bacterial diversity and richness between the healthy and diarrheal piglets. Principal coordinates analysis (PCoA) showed structural segregation between diseased and healthy groups, as well as among 3 different age groups. The abundance of , , , and increased due to dysbiosis induced by PEDV infection. Notably, there was a remarkable age-related increase in and in diarrheal piglets. Certain SCFA-producing bacteria, such as , , and , were shared by all healthy piglets, but were not identified in various age groups of diarrheal piglets. In addition, significant differences were observed between clusters of orthologous groups (COG) functional categories of healthy and PEDV-infected piglets. Our findings demonstrated that PEDV infection caused severe perturbations in porcine gut microbiota. Therefore, regulating gut microbiota in an age-related manner may be a promising method for the prevention or treatment of PEDV.
猪流行性腹泻(PED)是一种对家畜具有毁灭性影响的疾病。目前,大多数研究集中在比较健康仔猪和患PED仔猪的肠道微生物群,导致与腹泻仔猪动态变化相关的肠道微生物种群了解不足。本研究分析了猪流行性腹泻病毒(PEDV)感染仔猪在哺乳过渡阶段的肠道微生物群特征。从同一养猪场收集了1至3周龄的健康仔猪(n = 20)和PEDV感染仔猪(n = 18)的新鲜粪便样本。从每个样本中提取总DNA,并使用Illumina MiSeq平台扩增和测序16S rRNA基因的V3-V4高变区。在健康仔猪和腹泻仔猪之间观察到细菌多样性和丰富度存在统计学上的显著差异。主坐标分析(PCoA)显示患病组和健康组之间以及3个不同年龄组之间存在结构分离。由于PEDV感染引起的生态失调,[具体菌属名称1]、[具体菌属名称2]、[具体菌属名称3]和[具体菌属名称4]的丰度增加。值得注意的是,腹泻仔猪中[具体菌属名称5]和[具体菌属名称6]与年龄相关的增加显著。某些产生短链脂肪酸的细菌,如[具体菌属名称7]、[具体菌属名称8]和[具体菌属名称9],在所有健康仔猪中都有,但在腹泻仔猪的各个年龄组中未被鉴定到。此外,在健康仔猪和PEDV感染仔猪的直系同源群(COG)功能类别簇之间观察到显著差异。我们的研究结果表明,PEDV感染导致猪肠道微生物群严重紊乱。因此,以年龄相关的方式调节肠道微生物群可能是预防或治疗PEDV的一种有前景的方法。