Division of Infection and Immunity, University College London, United Kingdom.
Great Ormond Street Hospital for Children, United Kingdom.
Clin Infect Dis. 2019 Oct 30;69(10):1649-1656. doi: 10.1093/cid/ciz020.
Influenza A virus causes annual epidemics in humans and is associated with significant morbidity and mortality. Haemagglutinin (HA) and neuraminidase (NA) gene sequencing have traditionally been used to identify the virus genotype, although their utility in detecting outbreak clusters is still unclear. The objective of this study was to determine the utility, if any, of whole-genome sequencing over HA/NA sequencing for infection prevention and control (IPC) in hospitals.
We obtained all clinical samples from influenza (H1N1)-positive patients at the Great Ormond Street Hospital between January and March 2016. Samples were sequenced using targeted enrichment on an Illumina MiSeq sequencer. Maximum likelihood trees were computed for both whole genomes and concatenated HA/NA sequences. Epidemiological data was taken from routine IPC team activity during the period.
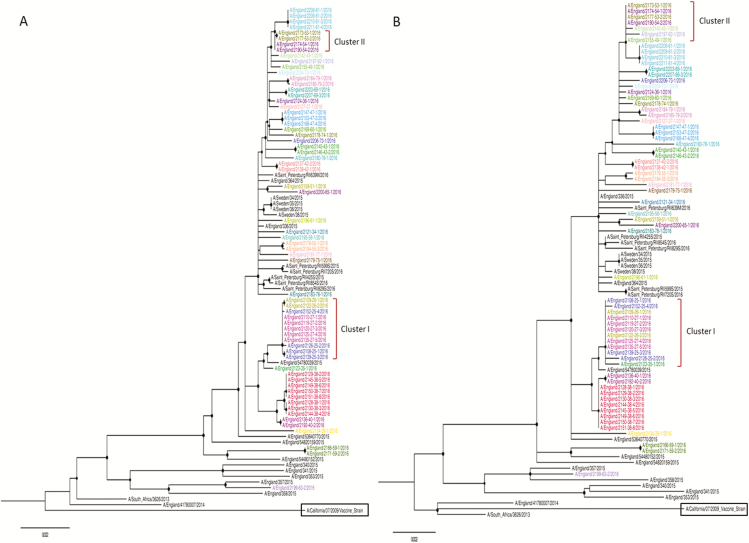
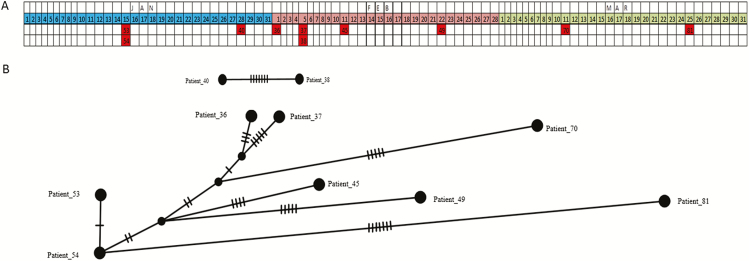
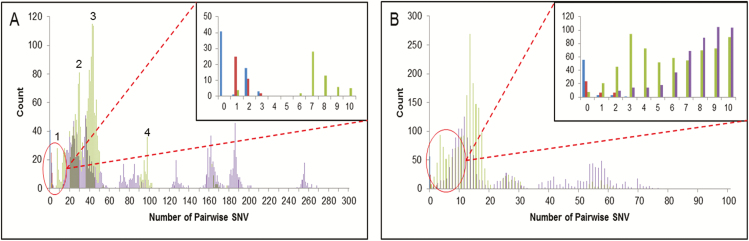
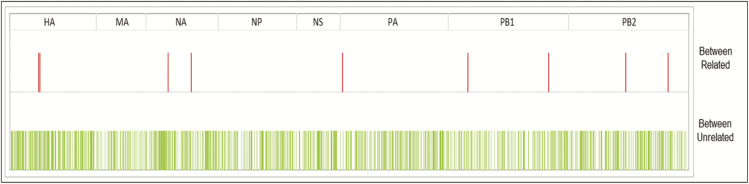
Complete genomes were obtained for 65/80 samples from 38 patients. Conventional IPC analysis recognized 1 outbreak, involving 3 children, and identified another potential cluster in the haemato-oncology ward. Whole-genome and HA/NA phylogeny both accurately identified the previously known outbreak cluster. However, HA/NA sequencing additionally identified unrelated strains as part of this outbreak cluster. A whole-genome analysis identified a further cluster of 2 infections that had been previously missed and refuted suspicions of transmission in the haemato-oncology wards.
Whole-genome sequencing is better at identifying outbreak clusters in a hospital setting than HA/NA sequencing. Whole-genome sequencing could provide a faster and more reliable method for outbreak monitoring and supplement routine IPC team work to allow the prevention of transmission.
甲型流感病毒每年在人类中引发流行,并导致重大发病率和死亡率。血凝素(HA)和神经氨酸酶(NA)基因测序传统上用于鉴定病毒基因型,尽管其在检测暴发集群中的作用仍不清楚。本研究的目的是确定全基因组测序在医院感染预防和控制(IPC)中的任何用途,是否优于 HA/NA 测序。
我们从 2016 年 1 月至 3 月在大奥蒙德街医院感染(H1N1)阳性患者中获得了所有临床样本。使用 Illumina MiSeq 测序仪进行靶向富集对样本进行测序。为全基因组和连接的 HA/NA 序列计算最大似然树。从该期间常规 IPC 团队活动中获取流行病学数据。
从 38 名患者的 80 个样本中获得了 65 个完整基因组。常规 IPC 分析识别出了 1 起暴发事件,涉及 3 名儿童,并在血液肿瘤病房中发现了另一个潜在的集群。全基因组和 HA/NA 系统发育都准确地鉴定了已知的暴发集群。但是,HA/NA 测序还将其他不相关的菌株鉴定为暴发集群的一部分。全基因组分析鉴定了另外 2 例先前漏检的感染,并且驳斥了血液肿瘤病房中传播的怀疑。
全基因组测序在医院环境中比 HA/NA 测序更能识别暴发集群。全基因组测序可以为暴发监测提供更快、更可靠的方法,并补充常规 IPC 团队的工作,以防止传播。