Department of Neurophysiology, Brain Research Institute, Niigata University, Niigata, 951-8585, Japan.
Department of Anesthesiology, Faculty of Medicine, Niigata University, Niigata, 951-8510, Japan.
J Physiol. 2019 Jul;597(13):3441-3455. doi: 10.1113/JP277615. Epub 2019 May 28.
Neuropathic pain spreads spatially beyond the injured sites, and the mechanism underlying the spread has been attributed to inflammation occurring in the spinal cord. However, the spatial spread of spinal/cortical potentiation induced by conduction block of the peripheral nerves can be observed prior to inflammation. In the present study, we found that spreading potentiation and hypersensitivity acutely induced by unilateral hindpaw ischaemia are nitric oxide (NO)-dependent and that NO is produced by ischaemia and quickly diffuses within the spinal cord. We also found that NO production induced by ischaemia is not observed in the presence of an antagonist for group II metabotropic glutamate receptors (mGluRs) and that neuronal NO synthase-positive dorsal horn neurons express group II mGluRs. These results suggest strongly that NO-mediated spreading potentiation in the spinal cord is one of the trigger mechanisms for neuropathic pain.
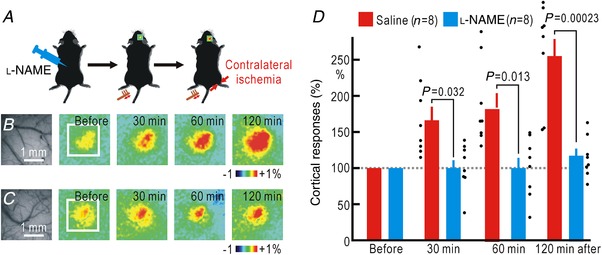
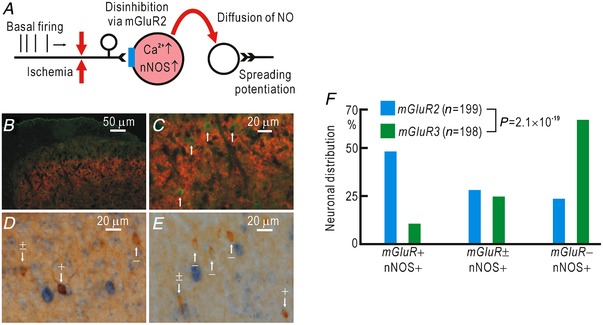
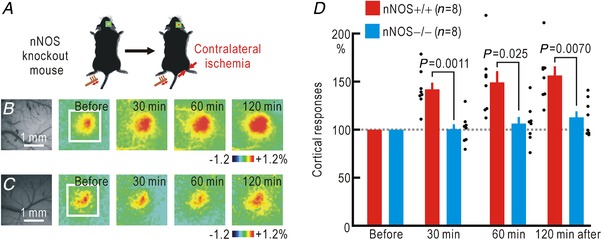
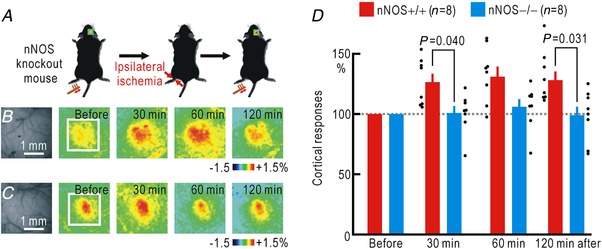
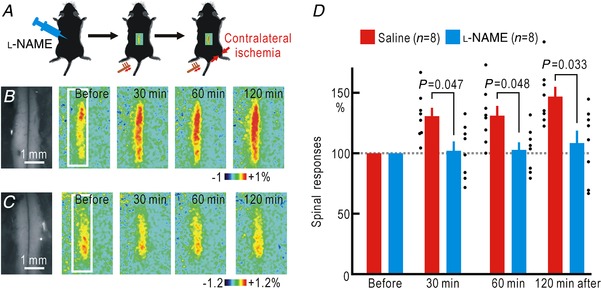
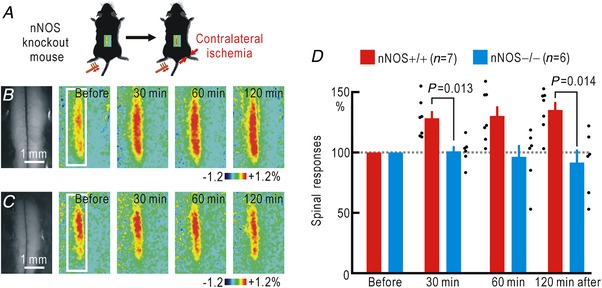
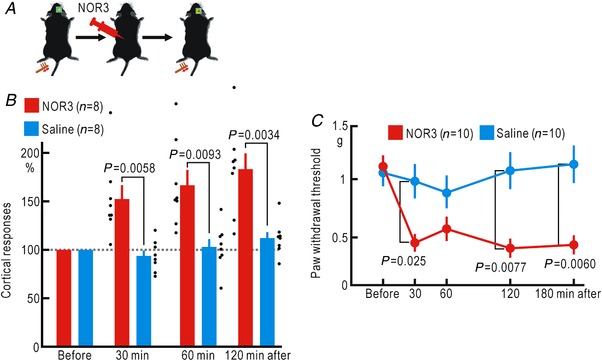
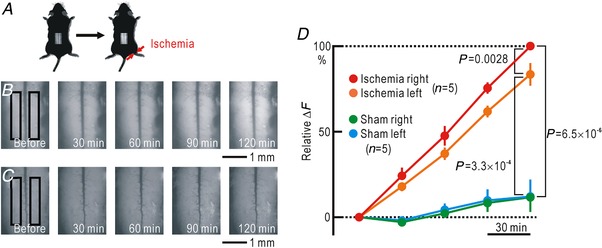
Cortical/spinal responses to hindpaw stimulation are bilaterally potentiated by unilateral hindpaw ischaemia in mice. We tested the hypothesis that hindpaw ischaemia produces nitric oxide (NO), which diffuses in the spinal cord to induce spatially spreading potentiation. Using flavoprotein fluorescence imaging, we confirmed that the spreading potentiation in hindpaw responses was induced during ischaemia in the non-stimulated hindpaw. This spreading potentiation was blocked by spinal application of l-NAME, an inhibitor of NO synthase (NOS). Furthermore, no spreading potentiation was observed in neural NOS (nNOS) knockout mice. Spinal application of an NO donor was enough to induce cortical potentiation and mechanical hypersensitivity. The spatial distribution of NO during unilateral hindpaw ischaemia was visualized using 4-amino-5-methylamino-2',7'-difluorofluorescein (DAF-FM). An increase in fluorescence derived from the complex of DAF-FM with NO was observed on the ischaemic side of the spinal cord. A similar but smaller increase was also observed on the contralateral side. Somatosensory potentiation after hindpaw ischaemia is known to be inhibited by spinal application of LY354740, an agonist of group II metabotropic glutamate receptors (mGluRs). We confirmed that the spinal DAF-FM fluorescence increases during hindpaw ischaemia were not observed in the presence of LY354740. We also confirmed that approximately half of the nNOS-positive neurons in the superficial laminae of the dorsal horn expressed mGluR2 mRNA. These results suggest that disinhibition of mGluR2 produces NO which in turn induces a spreading potentiation in a wide area of the spinal cord. Such spreading, along with the consequent non-specific potentiation in the spinal cord, may trigger neuropathic pain.
神经病理性疼痛在空间上超出损伤部位扩散,而这种扩散的机制归因于脊髓中发生的炎症。然而,外周神经传导阻滞诱导的脊髓/皮质增强的空间扩散可以在炎症之前观察到。在本研究中,我们发现单侧后爪缺血引起的扩散增强和超敏反应是一氧化氮(NO)依赖性的,并且缺血迅速产生 NO 并在脊髓内扩散。我们还发现,在存在代谢型谷氨酸受体(mGluR)II 型拮抗剂的情况下,不会观察到缺血诱导的 NO 产生,并且神经元型一氧化氮合酶阳性背角神经元表达 mGluR2。这些结果强烈表明,脊髓中的 NO 介导的扩散增强是神经病理性疼痛的触发机制之一。
在小鼠中,单侧后爪缺血会使双侧后爪刺激的皮质/脊髓反应增强。我们检验了这样的假设,即后爪缺血会产生一氧化氮(NO),NO 在脊髓内扩散以诱导空间扩散增强。使用黄素蛋白荧光成像,我们证实了在非刺激后爪的缺血过程中,后爪反应的扩散增强。这种扩散增强被脊髓应用一氧化氮合酶(NOS)抑制剂 l-NAME 阻断。此外,在神经型一氧化氮合酶(nNOS)敲除小鼠中,未观察到扩散增强。脊髓应用一氧化氮供体足以诱导皮质增强和机械性超敏反应。使用 4-氨基-5-甲基氨基-2',7'-二氟荧光素(DAF-FM)可视化单侧后爪缺血过程中的 NO 空间分布。在脊髓的缺血侧观察到与 DAF-FM 与 NO 复合物相关的荧光增加。在对侧也观察到类似但较小的增加。已知后爪缺血后的感觉敏化可被脊髓应用 mGluR2 代谢型谷氨酸受体(mGluR)激动剂 LY354740 抑制。我们证实,在 LY354740 存在的情况下,后爪缺血过程中脊髓内 DAF-FM 荧光的增加不会被观察到。我们还证实,背角浅层中约一半的 nNOS 阳性神经元表达 mGluR2 mRNA。这些结果表明,mGluR2 的去抑制作用产生 NO,NO 进而在脊髓的广泛区域诱导扩散增强。这种扩散以及脊髓内的非特异性增强,可能触发神经病理性疼痛。