Department of Pathology, Theriogenology, and One Health, Sao Paulo State University (FCAV-Unesp), Jaboticabal, Brazil.
Department of Preventive Veterinary Medicine, College of Veterinary Medicine, The Ohio State University, Columbus, OH, United States.
Front Cell Infect Microbiol. 2022 Jun 20;12:772829. doi: 10.3389/fcimb.2022.772829. eCollection 2022.
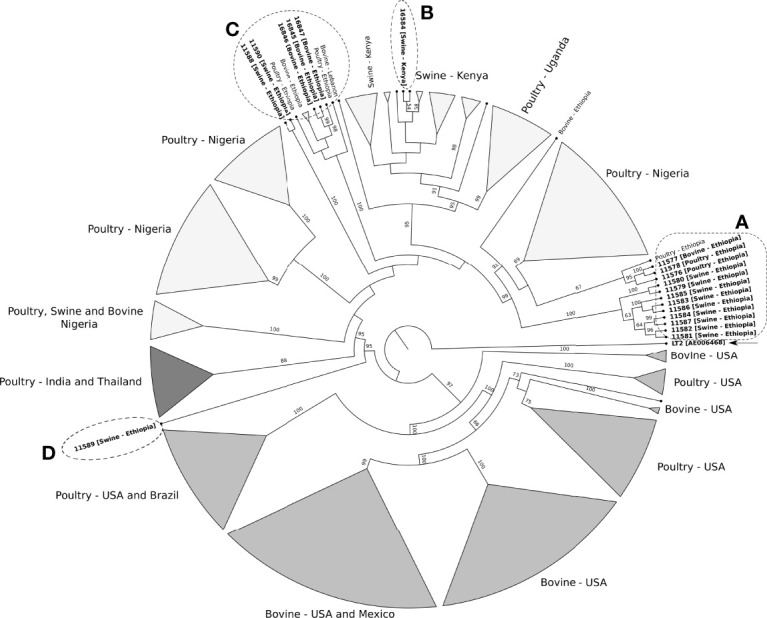
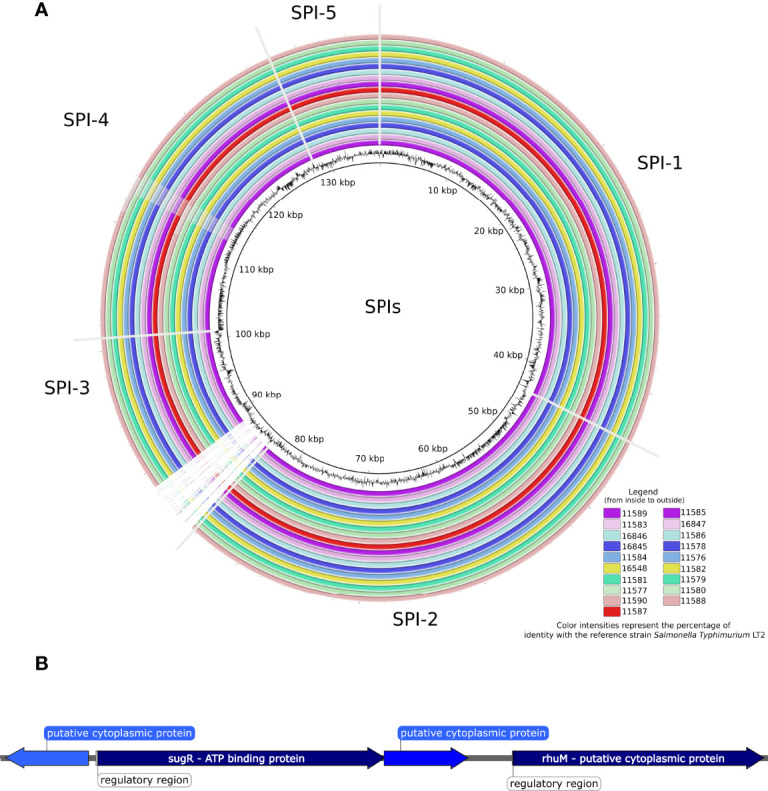
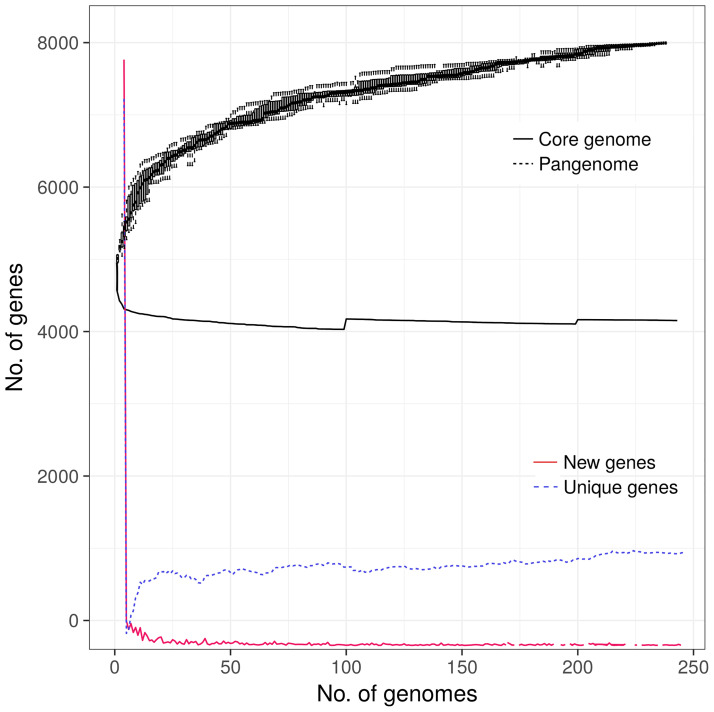
Since its emergence in the beginning of the 90's, multidrug-resistant (MDR) subsp serovar Kentucky has become a significant public health problem, especially in East Africa. This study aimed to investigate the antimicrobial resistance profile and the genotypic relatedness of Kentucky isolated from animal sources in Ethiopia and Kenya (n=19). We also investigated population evolutionary dynamics through phylogenetic and pangenome analyses with additional publicly available Kentucky ST198 genomes (n=229). All the 19 sequenced Kentucky isolates were identified as ST198. Among these isolates, the predominant genotypic antimicrobial resistance profile observed in ten (59.7%) isolates included the , , -, , , and (A) genes, which mediated resistance to gentamicin, streptomycin/spectinomycin, streptomycin, ampicillin, sulfamethoxazole and tetracycline, respectively; and A and C mutations associated to ciprofloxacin resistance. Four isolates harbored plasmid types Incl1 and/or Col8282; two of them carried both plasmids. Pathogenicity islands (SPI-1 to SPI-5) were highly conserved in the 19 sequenced Kentucky isolates. Moreover, at least one Pathogenicity Island (SPI 1-4, SPI 9 or C63PI) was identified among the 229 public Kentucky genomes. The phylogenetic analysis revealed that almost all Kentucky ST198 isolates (17/19) stemmed from a single strain that has accumulated ciprofloxacin resistance-mediating mutations. A total of 8,104 different genes were identified in a heterogenic and still open Kentucky ST198 pangenome. Considering the virulence factors and antimicrobial resistance genes detected in Kentucky, the implications of this pathogen to public health and the epidemiological drivers for its dissemination must be investigated.
自 90 年代初出现以来,多药耐药(MDR)亚种肯塔基州已成为一个重大的公共卫生问题,尤其是在东非。本研究旨在调查来自埃塞俄比亚和肯尼亚动物源的肯塔基州分离株的抗菌药物耐药谱和基因型相关性(n=19)。我们还通过与额外的公开可用的肯塔基州 ST198 基因组(n=229)进行系统发育和泛基因组分析,研究了种群进化动态。19 个测序的肯塔基州分离株均被鉴定为 ST198。在这些分离株中,十种(59.7%)分离株中观察到的主要基因型抗菌药物耐药谱包括介导对庆大霉素、链霉素/壮观霉素、链霉素、氨苄西林、磺胺甲噁唑和四环素耐药的 、 、 、 、和 (A)基因;以及与环丙沙星耐药相关的 A 和 C 突变。四个分离株携带 Inc1 型和/或 Col8282 型质粒;其中两个携带两种质粒。19 个测序的肯塔基州分离株中高度保守的致病岛(SPI-1 至 SPI-5)。此外,在 229 个公开的肯塔基州基因组中,至少有一个致病岛(SPI 1-4、SPI 9 或 C63PI)被鉴定出来。系统发育分析显示,几乎所有肯塔基州 ST198 分离株(17/19)都源自一株积累了介导环丙沙星耐药的突变的单一菌株。在一个异质且仍未关闭的肯塔基州 ST198 泛基因组中,共鉴定出 8104 个不同的基因。考虑到肯塔基州检测到的毒力因子和抗菌药物耐药基因,必须调查该病原体对公共卫生的影响及其传播的流行病学驱动因素。