Department of Entomology and Plant Pathology, North Carolina State University, Raleigh, United States.
Bioinformatics Research Center, North Carolina State University, Raleigh, United States.
Elife. 2019 Aug 15;8:e45562. doi: 10.7554/eLife.45562.
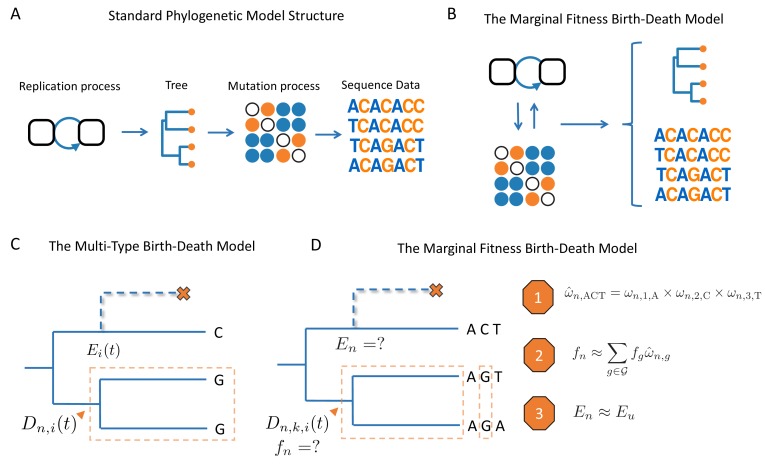
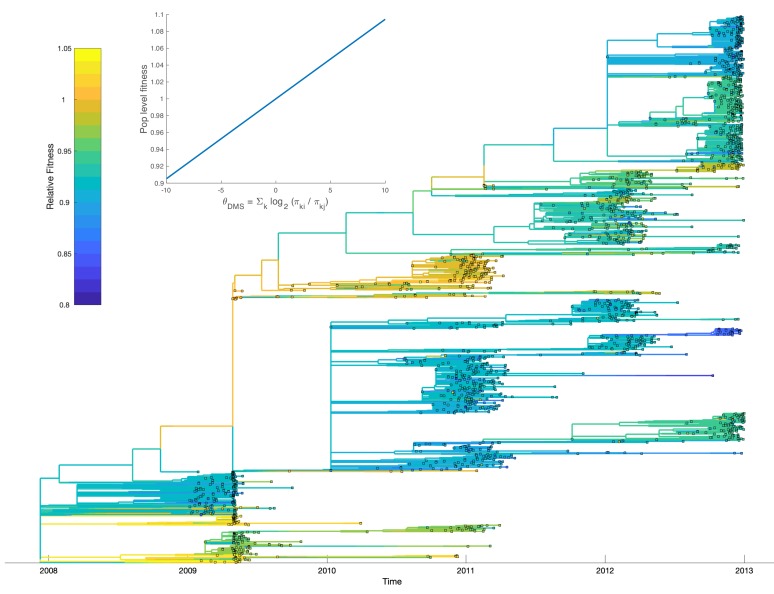
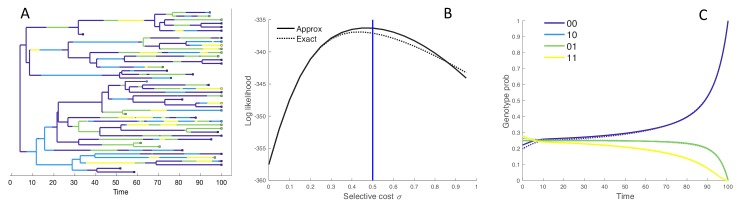
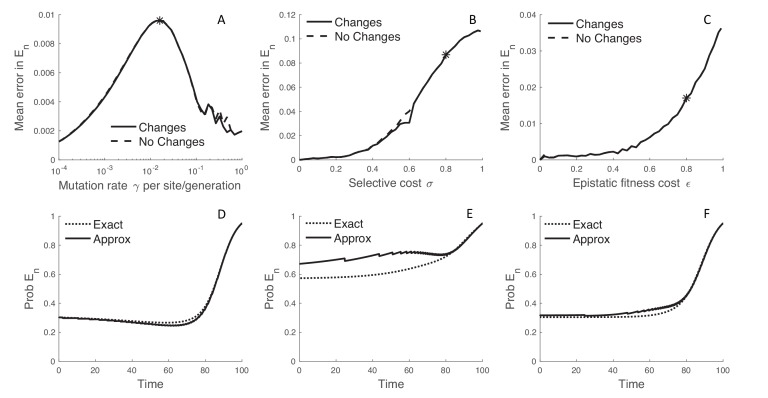
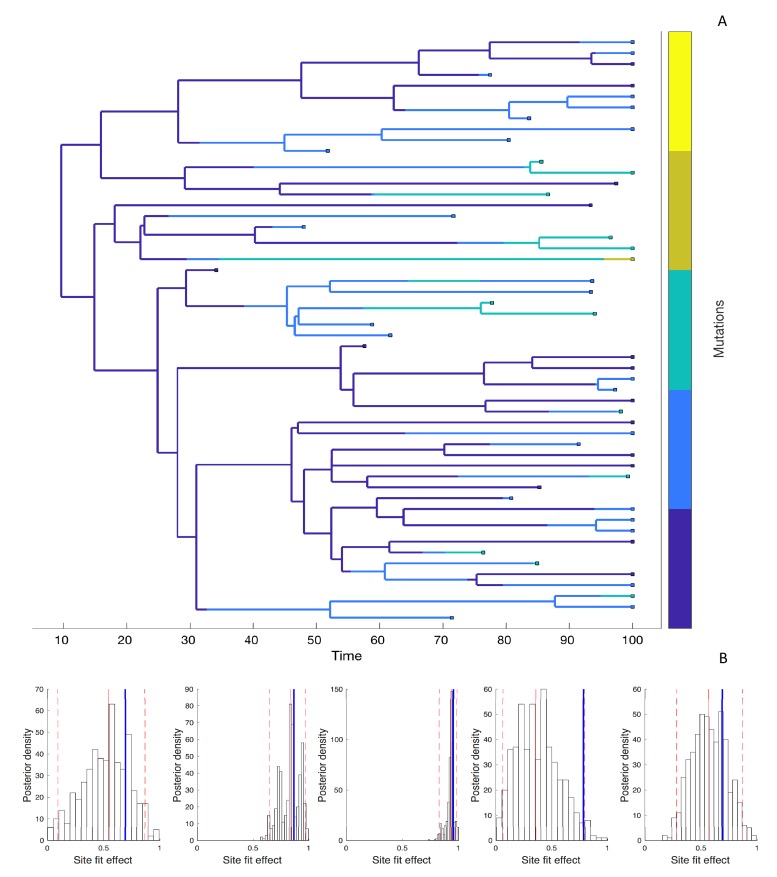
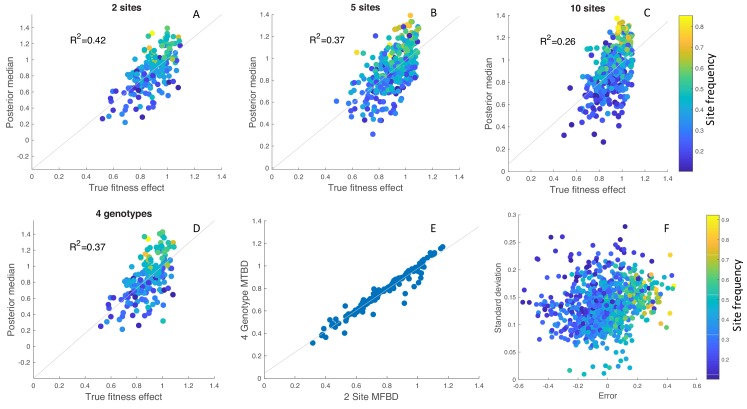
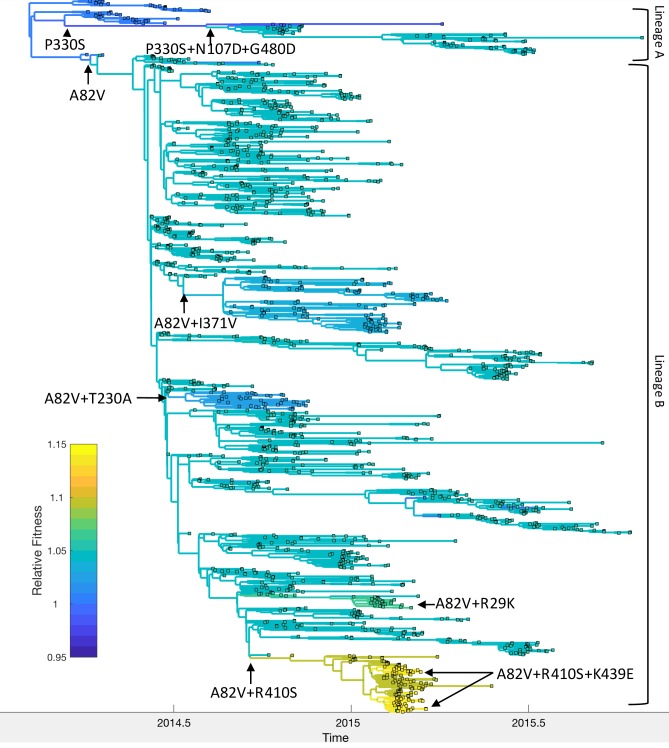
Beneficial and deleterious mutations cause the fitness of lineages to vary across a phylogeny and thereby shape its branching structure. While standard phylogenetic models do not allow mutations to feedback and shape trees, birth-death models can account for this feedback by letting the fitness of lineages depend on their type. To date, these multi-type birth-death models have only been applied to cases where a lineage's fitness is determined by a single character state. We extend these models to track sequence evolution at multiple sites. This approach remains computationally tractable by tracking the genotype and fitness of lineages probabilistically in an approximate manner. Although approximate, we show that we can accurately estimate the fitness of lineages and site-specific mutational fitness effects from phylogenies. We apply this approach to estimate the population-level fitness effects of mutations in Ebola and influenza virus, and compare our estimates with in vitro fitness measurements for these mutations.
有益突变和有害突变会导致谱系在系统发育上的适应性发生变化,从而塑造其分支结构。虽然标准的系统发育模型不允许突变进行反馈和塑造树,但出生-死亡模型可以通过让谱系的适应性取决于它们的类型来解释这种反馈。迄今为止,这些多类型出生-死亡模型仅应用于谱系适应性由单个特征状态决定的情况。我们将这些模型扩展到可以在多个位点跟踪序列进化的情况。通过以近似的方式概率跟踪谱系的基因型和适应性,这种方法仍然具有计算可行性。虽然是近似的,但我们表明,我们可以从系统发育中准确估计谱系的适应性和位点特异性突变适应性效应。我们将这种方法应用于估计埃博拉病毒和流感病毒突变的群体适应性效应,并将我们的估计与这些突变的体外适应性测量值进行比较。