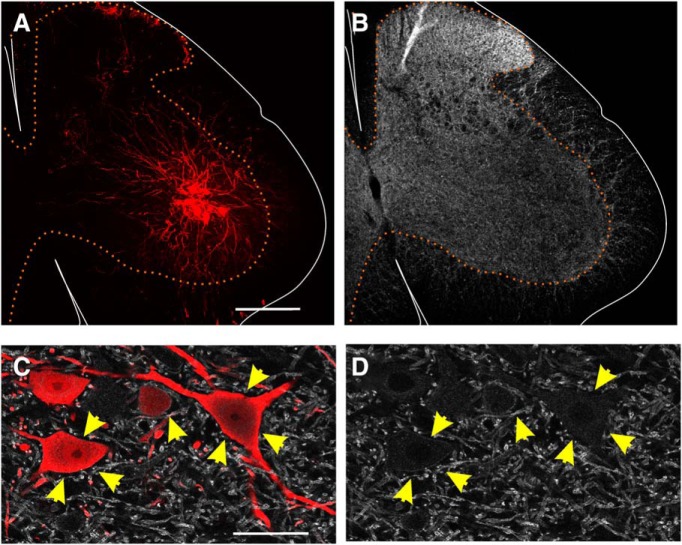
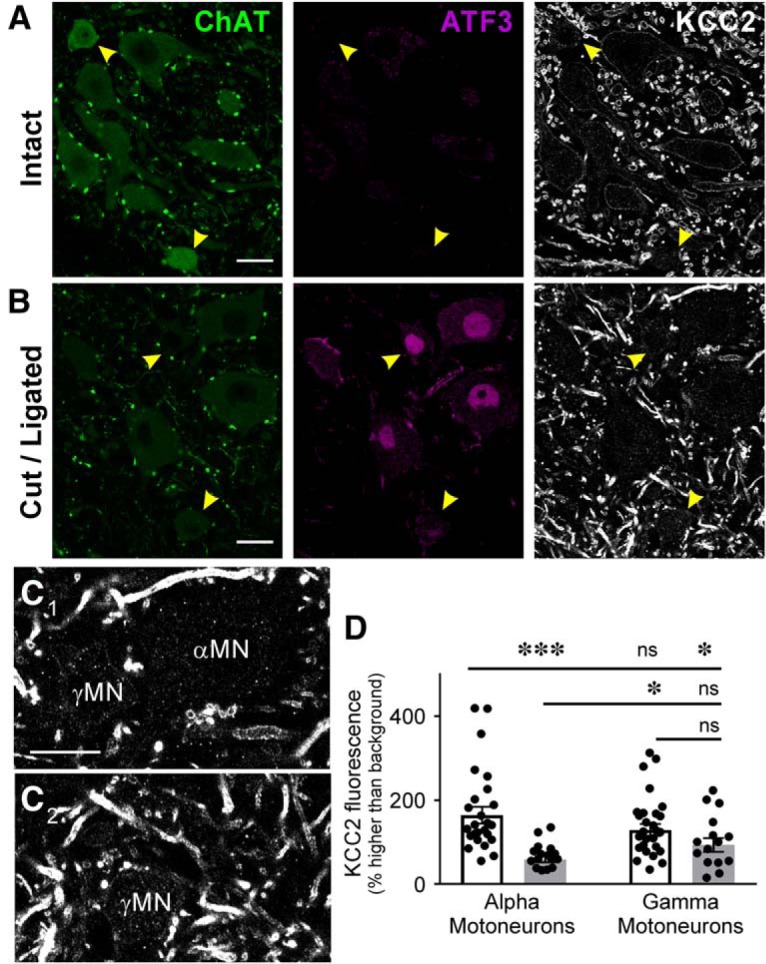
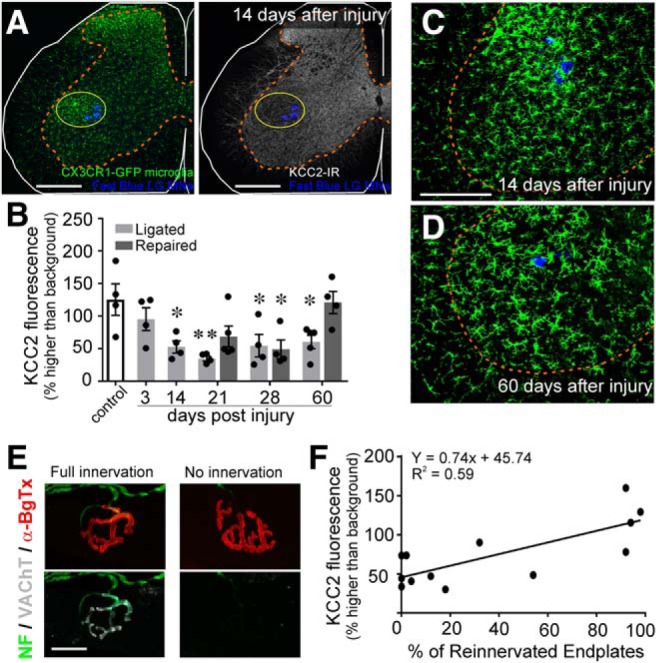
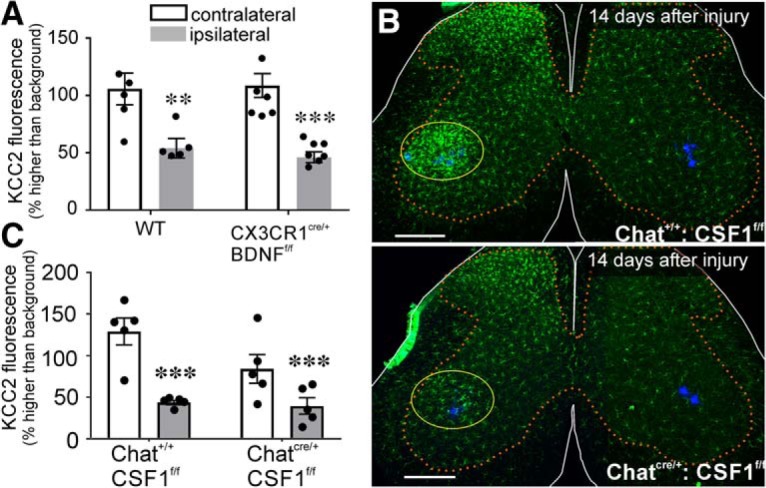
Departments of Physiology, Emory University, Atlanta, GA 30322.
Cell Biology, Emory University, Atlanta, GA 30322.
eNeuro. 2019 Oct 16;6(5). doi: 10.1523/ENEURO.0172-19.2019. Print 2019 Sep/Oct.
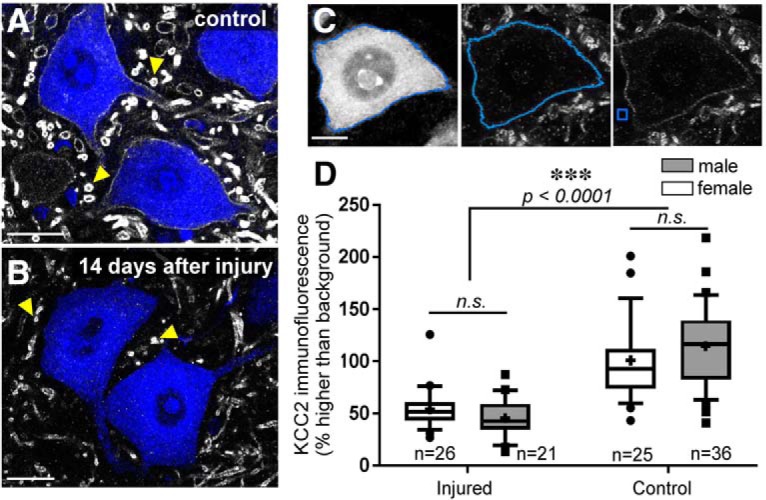
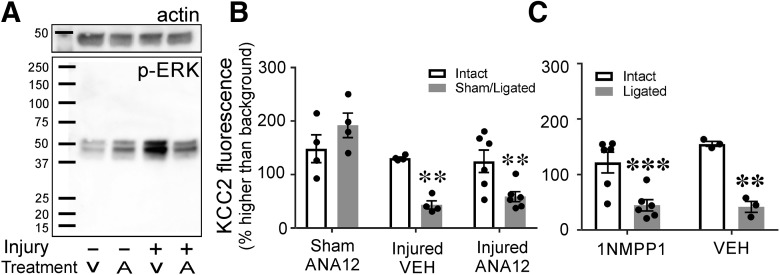
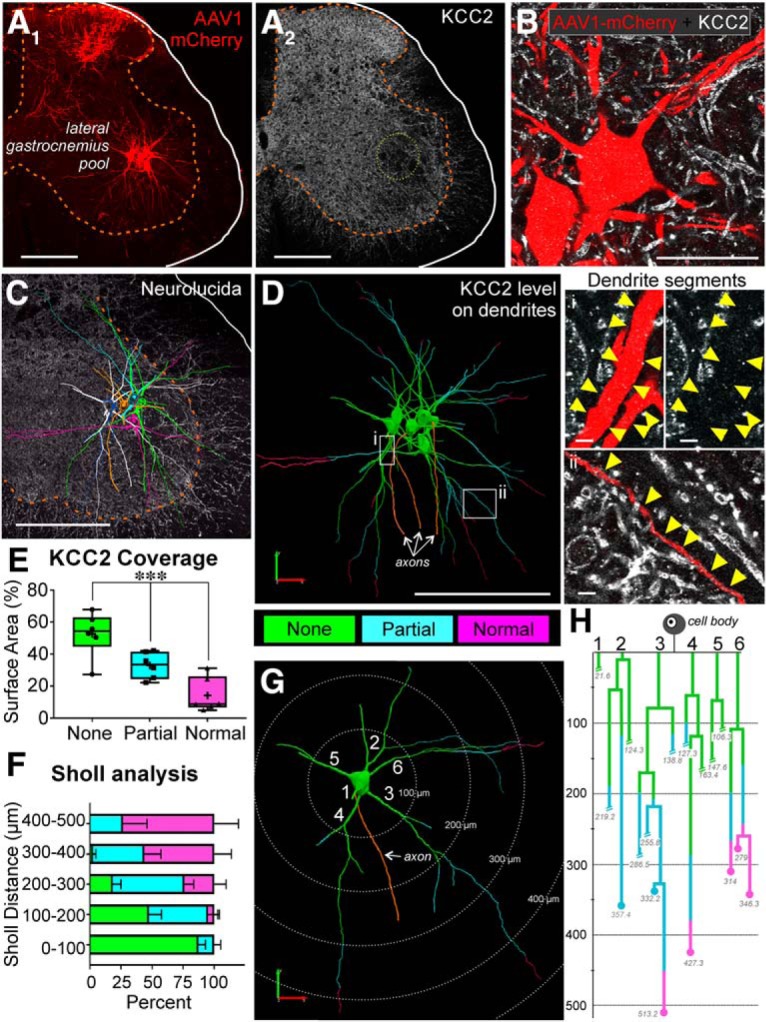
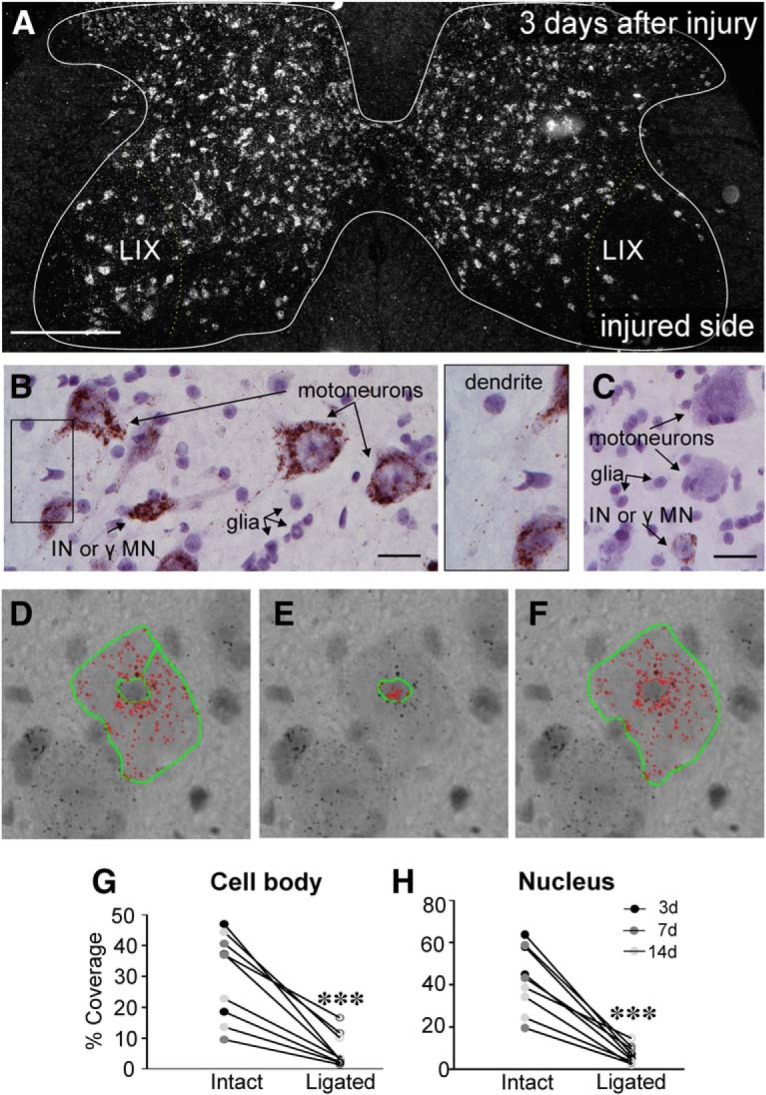
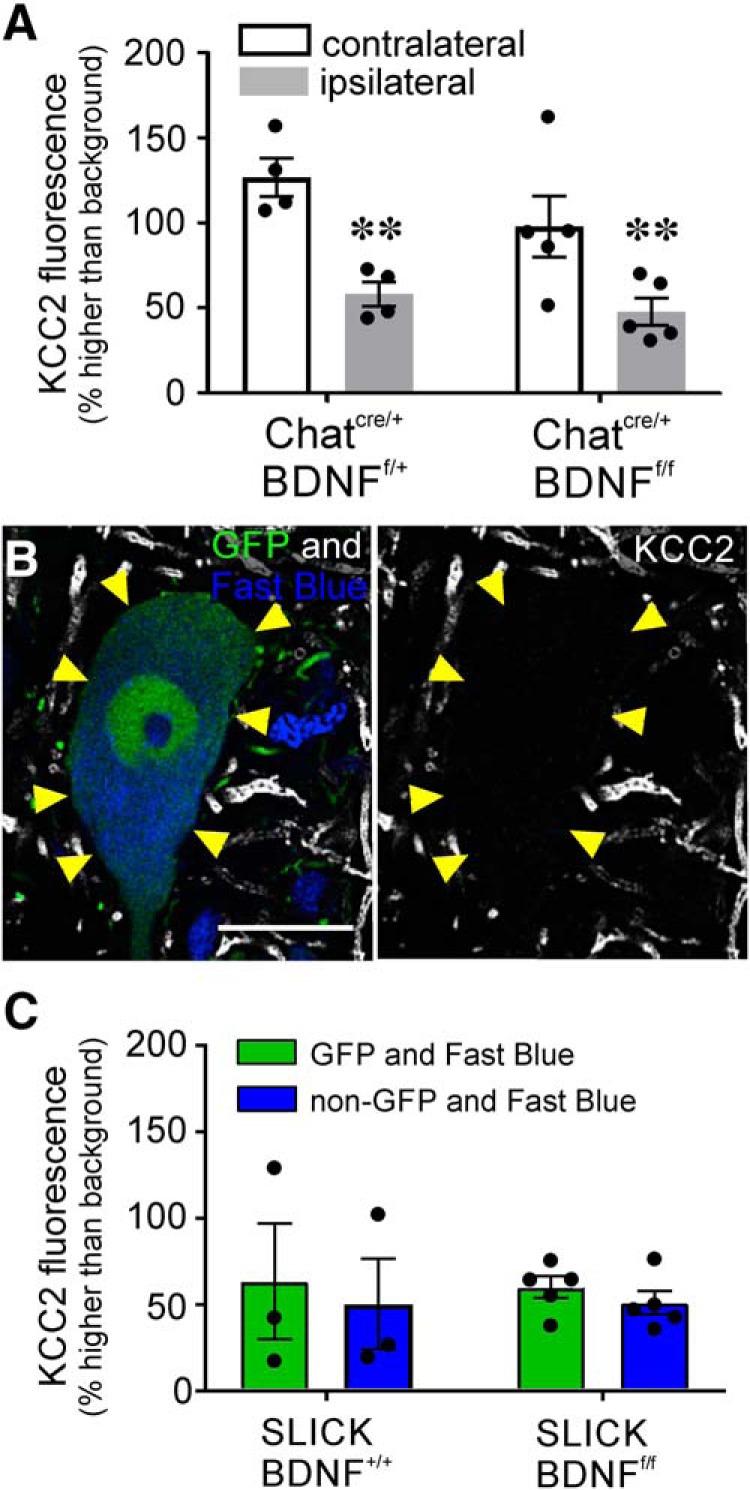
The potassium-chloride cotransporter (KCC2) maintains the low intracellular chloride found in mature central neurons and controls the strength and direction of GABA/glycine synapses. We found that following axotomy as a consequence of peripheral nerve injuries (PNIs), KCC2 protein is lost throughout the somatodendritic membrane of axotomized spinal cord motoneurons after downregulation of mRNA expression. This large loss likely depolarizes the reversal potential of GABA/glycine synapses, resulting in GABAergic-driven spontaneous activity in spinal motoneurons similar to previous reports in brainstem motoneurons. We hypothesized that the mechanism inducing KCC2 downregulation in spinal motoneurons following peripheral axotomy might be mediated by microglia or motoneuron release of BDNF and TrkB activation as has been reported on spinal cord dorsal horn neurons after nerve injury, motoneurons after spinal cord injury (SCI), and in many other central neurons throughout development or a variety of pathologies. To test this hypothesis, we used genetic approaches to interfere with microglia activation or delete from specifically microglia or motoneurons, as well as pharmacology (ANA-12) and pharmacogenetics (F616A mice) to block TrkB activation. We show that KCC2 dysregulation in axotomized motoneurons is independent of microglia, BDNF, and TrkB. KCC2 is instead dependent on neuromuscular innervation; KCC2 levels are restored only when motoneurons reinnervate muscle. Thus, downregulation of KCC2 occurs specifically while injured motoneurons are regenerating and might be controlled by target-derived signals. GABAergic and glycinergic synapses might therefore depolarize motoneurons disconnected from their targets and contribute to augment motoneuron activity known to promote motor axon regeneration.
钾氯共转运蛋白(KCC2)维持成熟中枢神经元中发现的低细胞内氯离子,并控制 GABA/甘氨酸突触的强度和方向。我们发现,在外周神经损伤(PNI)导致轴突切断后,KCC2 蛋白的 mRNA 表达下调后,整个轴突切断的脊髓运动神经元的体树突膜中丢失。这种大量的丢失很可能使 GABA/甘氨酸突触的反转电位去极化,导致脊髓运动神经元中 GABA 能驱动的自发性活动,类似于先前在脑干运动神经元中的报道。我们假设,外周轴突切断后脊髓运动神经元中 KCC2 下调的机制可能是由小胶质细胞或运动神经元释放 BDNF 和 TrkB 激活介导的,正如神经损伤后脊髓背角神经元、脊髓损伤后运动神经元以及许多其他发育过程中的中枢神经元或各种病理状态下的报道。为了验证这一假说,我们使用了基因方法干扰小胶质细胞的激活或特异性地从小胶质细胞或运动神经元中删除,以及药理学(ANA-12)和药物遗传学(F616A 小鼠)来阻断 TrkB 的激活。我们表明,轴突切断的运动神经元中 KCC2 的失调与小胶质细胞、BDNF 和 TrkB 无关。相反,KCC2 依赖于神经肌肉支配;只有当运动神经元重新支配肌肉时,KCC2 水平才会恢复。因此,KCC2 的下调仅发生在受损的运动神经元再生期间,并且可能受到靶源性信号的控制。因此,GABA 能和甘氨酸能突触可能使与靶标分离的运动神经元去极化,并有助于增强已知促进运动轴突再生的运动神经元活动。