Department of Theoretical Chemistry, Lund University, Chemical Centre, P.O. Box 124, SE-221 00 Lund, Sweden.
J Chem Theory Comput. 2020 Mar 10;16(3):1936-1952. doi: 10.1021/acs.jctc.9b01254. Epub 2020 Feb 14.
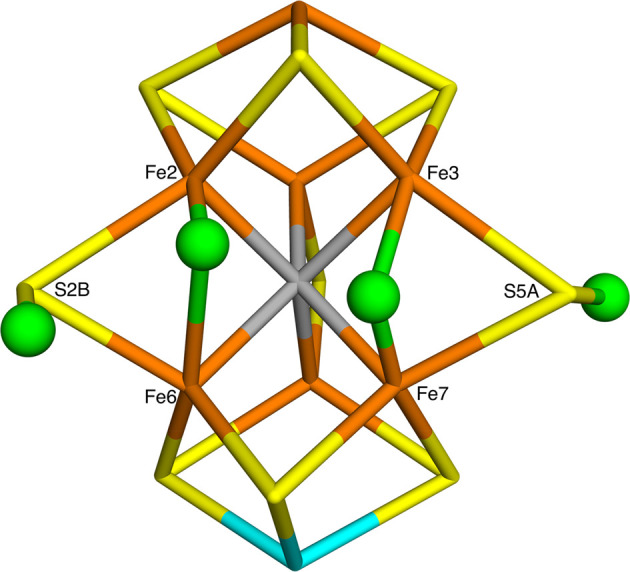
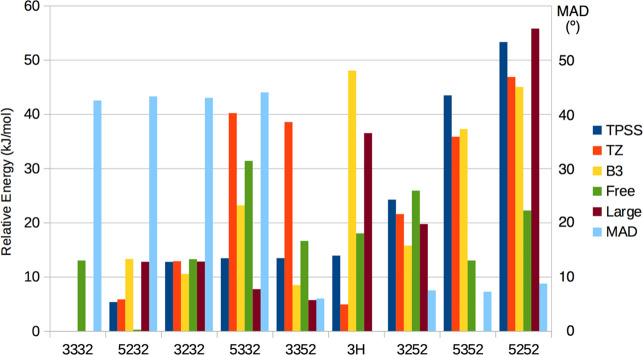
Nitrogenase is the only enzyme that can cleave the strong triple bond in N. The active site contains a complicated MoFeSC cluster. It is believed that it needs to accept four protons and electrons, forming the E state, before it can bind N. However, there is no consensus on the atomic structure of the E state. Experimental studies indicate that it should contain two hydride ions bridging two pairs of Fe ions, and it has been suggested that both hydride ions as well as the two protons bind on the same face of the cluster. On the other hand, density functional theory (DFT) studies have indicated that it is energetically more favorable with either three hydride ions or with a triply protonated carbide ion, depending on the DFT functional. We have performed a systematic combined quantum mechanical and molecular mechanical (QM/MM) study of possible E states with two bridging hydride ions. Our calculations suggest that the most favorable structure has hydride ions bridging the Fe2/6 and Fe3/7 ion pairs. In fact, such structures are 14 kJ/mol more stable than structures with three hydride ions, showing that pure DFT functionals give energetically most favorable structures in agreement with experiments. An important reason for this finding is that we have identified a new type of broken-symmetry state that involves only two Fe ions with minority spin, in contrast to the previously studied states with three Fe ions with minority spin. The energetically best structures have the two hydride ions on different faces of the FeMo cluster, whereas better agreement with ENDOR data is obtained if they are on the same face; such structures are only 6-22 kJ/mol less stable.
固氮酶是唯一能够切断 N 中强三键的酶。活性位点包含一个复杂的 MoFeSC 簇。据信,它需要接受四个质子和电子,形成 E 态,然后才能结合 N。然而,对于 E 态的原子结构还没有共识。实验研究表明,它应该包含两个桥连两个 Fe 离子对的氢化物离子,并且已经提出两个氢化物离子以及两个质子都结合在簇的同一面上。另一方面,密度泛函理论 (DFT) 研究表明,根据 DFT 函数,它更有利于存在三个氢化物离子或三重质子化的碳化物离子。我们已经对具有两个桥接氢化物离子的可能 E 态进行了系统的量子力学和分子力学 (QM/MM) 联合研究。我们的计算表明,最有利的结构是氢化物离子桥接 Fe2/6 和 Fe3/7 离子对。实际上,这种结构比具有三个氢化物离子的结构稳定 14 kJ/mol,表明纯 DFT 泛函与实验一致给出了最有利的能量结构。这一发现的一个重要原因是,我们已经确定了一种新的非对称破缺态,它只涉及两个具有少数自旋的 Fe 离子,与之前研究的具有三个具有少数自旋的 Fe 离子的态不同。能量最佳的结构是两个氢化物离子位于 FeMo 簇的不同面上,而与 ENDOR 数据的更好一致性是通过它们位于同一面上获得的;这种结构仅比能量最佳结构稳定 6-22 kJ/mol。