Department of Animal and Poultry Sciences, Virginia Polytechnic Institute and State University, Blacksburg, VA, United States of America.
Department of Agronomy and Horticulture, University of Nebraska, Lincoln, NE, United States of America.
PLoS One. 2020 Feb 3;15(2):e0228118. doi: 10.1371/journal.pone.0228118. eCollection 2020.
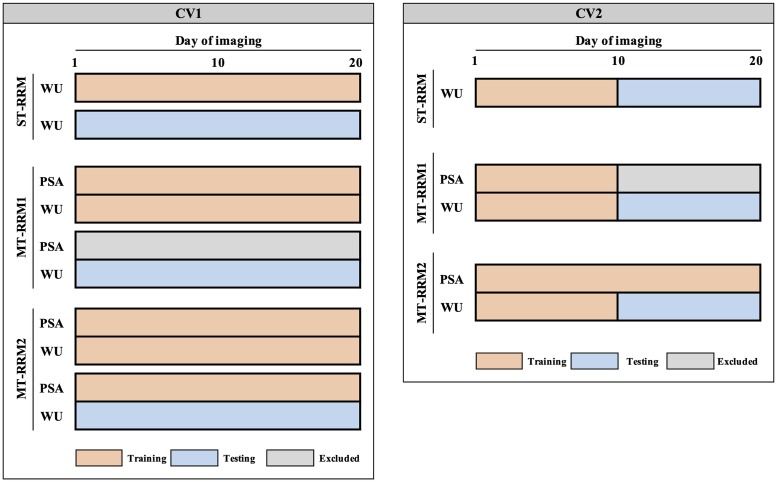
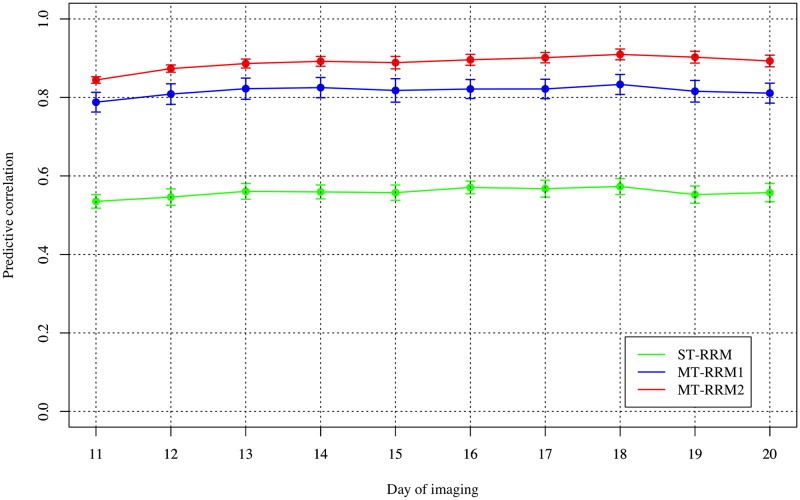
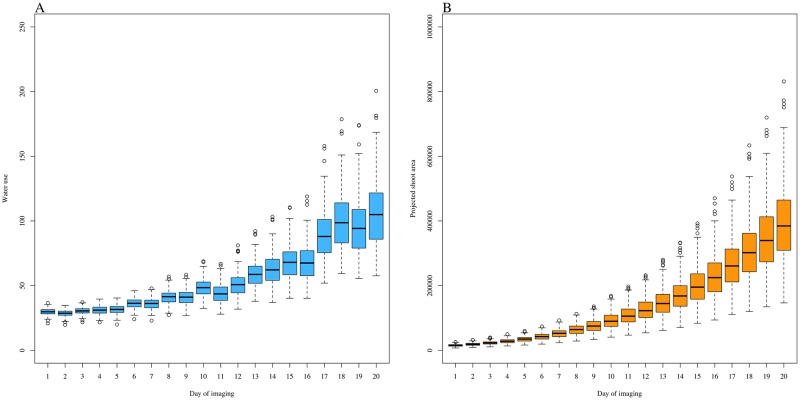
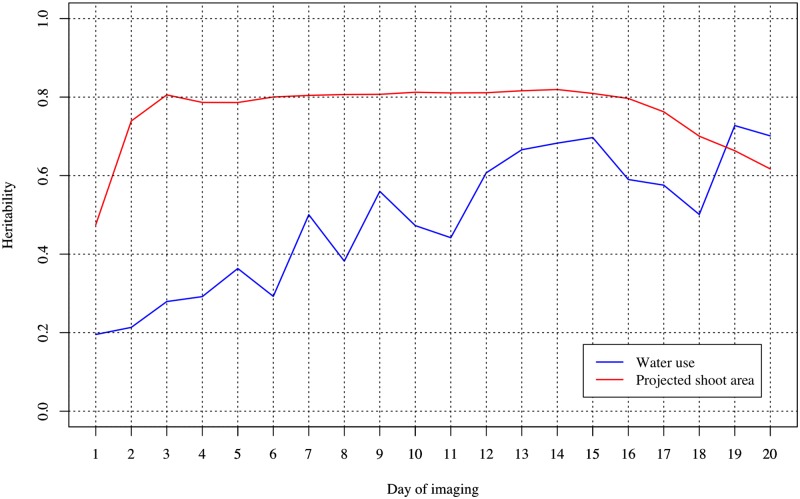
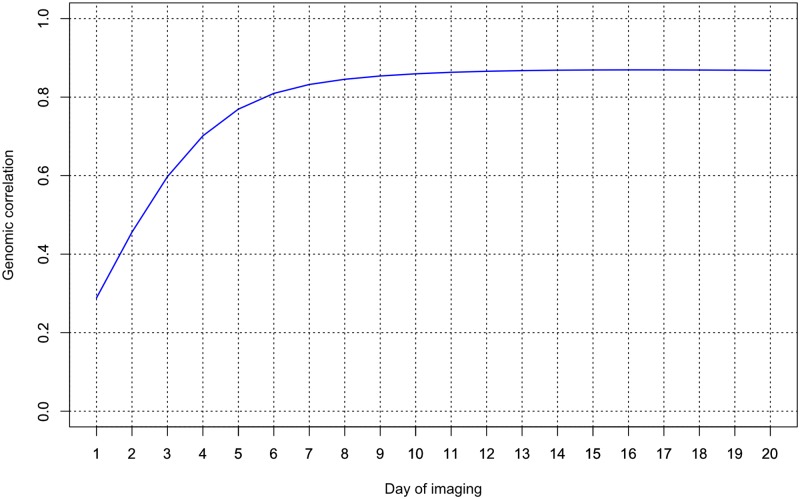
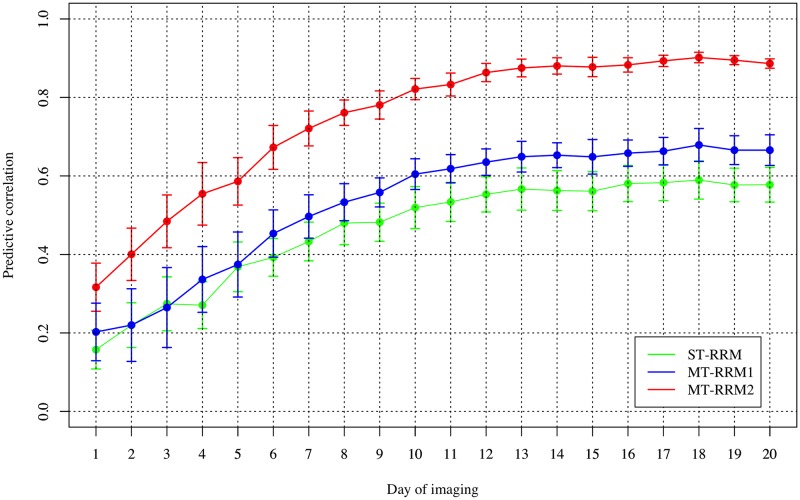
Random regression models (RRM) are used extensively for genomic inference and prediction of time-valued traits in animal breeding, but only recently have been used in plant systems. High-throughput phenotyping (HTP) platforms provide a powerful means to collect high-dimensional phenotypes throughout the growing season for large populations. However, to date, selection of an appropriate statistical genomic framework to integrate multiple temporal traits for genomic prediction in plants remains unexplored. Here, we demonstrate the utility of a multi-trait RRM (MT-RRM) for genomic prediction of daily water usage (WU) in rice (Oryza sativa) through joint modeling with shoot biomass (projected shoot area, PSA). Three hundred and fifty-seven accessions were phenotyped daily for WU and PSA over 20 days using a greenhouse-based HTP platform. MT-RRMs that modeled additive genetic and permanent environmental effects for both traits using quadratic Legendre polynomials were used to assess genomic correlations between traits and genomic prediction for WU. Predictive abilities of the MT-RRMs were assessed using two cross-validation (CV) scenarios. The first scenario was designed to predict genetic values for WU at all time points for a set of accessions with unobserved WU. The second scenario was designed to forecast future genetic values for WU for a panel of known accessions with records for WU at earlier time periods. In each scenario we evaluated two MT-RRMs in which PSA records were absent or available for time points in the testing population. Weak to strong genomic correlations between WU and PSA were observed across the days of imaging (0.29-0.870.38-0.80). In both CV scenarios, MT-RRMs showed better predictive abilities compared to single-trait RRM, and prediction accuracies were greatly improved when PSA records were available for the testing population. In summary, these frameworks provide an effective approach to predict temporal physiological traits that are difficult or expensive to quantify in large populations.
随机回归模型(RRM)广泛应用于动物育种中基因组推断和时间值性状的预测,但直到最近才在植物系统中使用。高通量表型(HTP)平台为在整个生长季节对大群体进行高维表型的收集提供了有力的手段。然而,迄今为止,选择适当的统计基因组框架来整合多个时间性状以进行植物的基因组预测仍然是一个未探索的问题。在这里,我们通过联合建模(将投影的 Shoot 面积 PSA 与每日耗水量 WU 进行建模)展示了多性状 RRM(MT-RRM)在水稻(Oryza sativa)基因组预测中对 WU 的应用。使用基于温室的 HTP 平台,对 357 个品系进行了 20 天的 WU 和 PSA 每日表型测定。MT-RRMs 使用二次勒让德多项式对两个性状的加性遗传和永久环境效应进行建模,用于评估性状间的基因组相关性和 WU 的基因组预测。使用两种交叉验证(CV)方案评估了 MT-RRMs 的预测能力。第一种方案旨在为一组未观察到 WU 的品系预测所有时间点的 WU 遗传值。第二种方案旨在为具有早期 WU 记录的已知品系面板预测未来的 WU 遗传值。在每种情况下,我们评估了两种 MT-RRMs,其中 PSA 记录在测试群体的时间点上缺失或可用。在成像的天数中观察到 WU 和 PSA 之间的弱到强的基因组相关性(0.29-0.87,0.38-0.80)。在两种 CV 方案中,MT-RRMs 与单性状 RRM 相比显示出更好的预测能力,并且当 PSA 记录可用于测试群体时,预测精度大大提高。总之,这些框架提供了一种有效的方法来预测在大群体中难以或昂贵地量化的时间生理性状。