Department of Computer Science, University of Toronto, Toronto, ON, Canada.
Vector Institute, Toronto, ON, Canada.
Nat Commun. 2020 Feb 5;11(1):731. doi: 10.1038/s41467-020-14352-7.
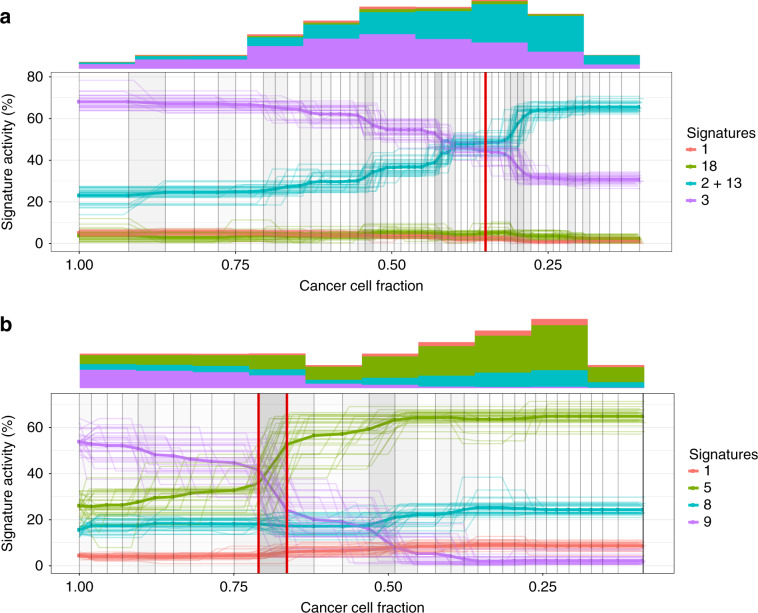
The type and genomic context of cancer mutations depend on their causes. These causes have been characterized using signatures that represent mutation types that co-occur in the same tumours. However, it remains unclear how mutation processes change during cancer evolution due to the lack of reliable methods to reconstruct evolutionary trajectories of mutational signature activity. Here, as part of the ICGC/TCGA Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium, which aggregated whole-genome sequencing data from 2658 cancers across 38 tumour types, we present TrackSig, a new method that reconstructs these trajectories using optimal, joint segmentation and deconvolution of mutation type and allele frequencies from a single tumour sample. In simulations, we find TrackSig has a 3-5% activity reconstruction error, and 12% false detection rate. It outperforms an aggressive baseline in situations with branching evolution, CNA gain, and neutral mutations. Applied to data from 2658 tumours and 38 cancer types, TrackSig permits pan-cancer insight into evolutionary changes in mutational processes.
癌症突变的类型和基因组背景取决于其成因。这些成因已通过代表在相同肿瘤中共同发生的突变类型的特征来描述。然而,由于缺乏可靠的方法来重建突变特征活动的进化轨迹,因此仍然不清楚由于癌症演变导致的突变过程如何发生变化。在这里,作为 ICGC/TCGA 全基因组泛癌症分析(PCAWG)联盟的一部分,该联盟聚合了来自 38 种肿瘤类型的 2658 种癌症的全基因组测序数据,我们提出了 TrackSig,这是一种使用来自单个肿瘤样本的突变类型和等位基因频率的最优联合分割和去卷积来重建这些轨迹的新方法。在模拟中,我们发现 TrackSig 的活动重建误差为 3-5%,假阳性检出率为 12%。在具有分支进化、CNA 增益和中性突变的情况下,它的表现优于激进基线。将 TrackSig 应用于来自 2658 个肿瘤和 38 种癌症类型的数据,可以全面了解癌症突变过程中的进化变化。