Libisch Balázs, Keresztény Tibor, Kerényi Zoltán, Kocsis Róbert, Sipos Rita, Papp Péter P, Olasz Ferenc
Laboratory of Microbiology, Agricultural Biotechnology Institute, National Agricultural Research and Innovation Centre (NARIC), 2100 Gödöllő, Hungary.
Hungarian Dairy Research Institute Ltd., 9200 Mosonmagyaróvár, Hungary.
J Vet Res. 2020 Feb 29;64(1):111-118. doi: 10.2478/jvetres-2020-0015. eCollection 2020 Mar.
Land application of manure that contains antibiotics and resistant bacteria may facilitate the establishment of an environmental reservoir of antibiotic-resistant microbes, promoting their dissemination into agricultural and natural habitats. The main objective of this study was to search for acquired antibiotic resistance determinants in the gut microbiota of wild boar populations living in natural habitats.
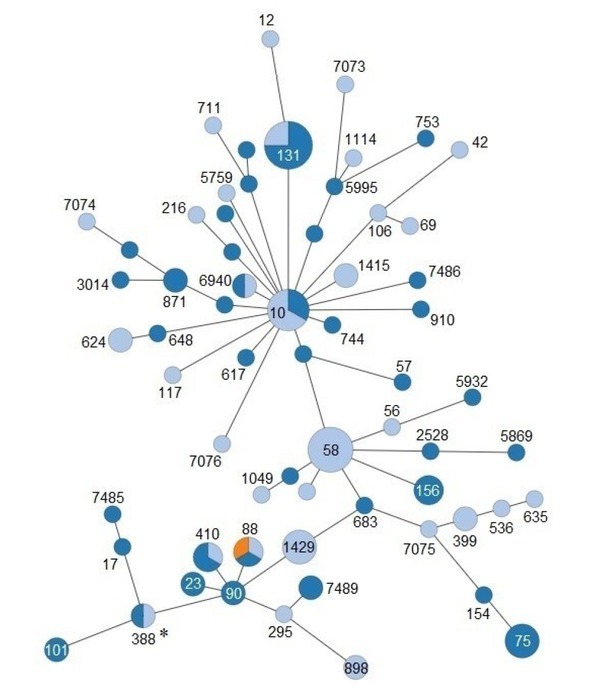
Gastrointestinal samples of free-living wild boars were collected in the Zemplén Mountains in Hungary and were characterised by culture-based, metagenomic, and molecular microbiological methods. Bioinformatic analysis of the faecal microbiome of a hunted wild boar from Japan was used for comparative studies. Also, shotgun metagenomic sequencing data of two untreated sewage wastewater samples from North Pest (Hungary) from 2016 were analysed by bioinformatic methods. Minimum spanning tree diagrams for seven-gene MLST profiles of 104 strains isolated in Europe from wild boars and domestic pigs were generated in Enterobase.
In the ileum of a diarrhoeic boar, a dominant O112ab:H2 strain with intermediate resistance to gentamicin, tobramycin, and amikacin was identified, displaying sequence type ST388 and harbouring the EAST1 toxin gene. Metagenomic analyses of the colon and rectum digesta revealed the presence of the , , , and antibiotic resistance genes that were also detected in the gut microbiome of four other wild boars from the mountains. Furthermore, the and genes were identified in the faecal microbiome of a hunted wild boar from Japan.
The gastrointestinal microbiota of the free-living wild boars examined in this study carried acquired antibiotic resistance determinants that are highly prevalent among domestic livestock populations.
施用含有抗生素和耐药菌的粪肥可能会促进环境中抗生素耐药微生物库的形成,促使它们传播到农业和自然栖息地。本研究的主要目的是在生活于自然栖息地的野猪种群的肠道微生物群中寻找获得性抗生素耐药决定因素。
在匈牙利的曾普伦山脉采集自由生活野猪的胃肠道样本,并采用基于培养、宏基因组学和分子微生物学方法进行特征分析。对一头来自日本的被猎杀野猪的粪便微生物群进行生物信息学分析以作比较研究。此外,还采用生物信息学方法分析了2016年来自匈牙利北佩斯的两个未经处理的污水废水样本的鸟枪法宏基因组测序数据。在Enterobase中生成了从欧洲野猪和家猪分离出的104株菌株的七基因多位点序列分型(MLST)图谱的最小生成树图。
在一头腹泻野猪的回肠中,鉴定出一株对庆大霉素、妥布霉素和阿米卡星具有中度耐药性的优势O112ab:H2菌株,其序列型为ST388,并携带EAST1毒素基因。对结肠和直肠消化物的宏基因组分析显示存在blaCTX-M-1、blaTEM-1、blaSHV-1和tet(A)抗生素耐药基因,这些基因也在来自山区的其他4头野猪的肠道微生物群中检测到。此外,在一头来自日本的被猎杀野猪的粪便微生物群中鉴定出blaNDM-1和mcr-1基因。
本研究中检测的自由生活野猪的胃肠道微生物群携带获得性抗生素耐药决定因素,这些因素在家畜种群中高度普遍。