TBI, Université de Toulouse, CNRS, INRAE, INSA, Toulouse, France.
Scientific IT Services, ETH Zurich, Zurich, Switzerland.
PLoS Comput Biol. 2020 Apr 14;16(4):e1007799. doi: 10.1371/journal.pcbi.1007799. eCollection 2020 Apr.
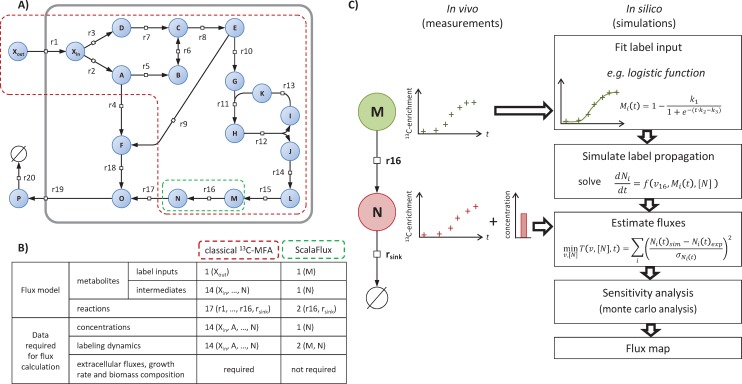
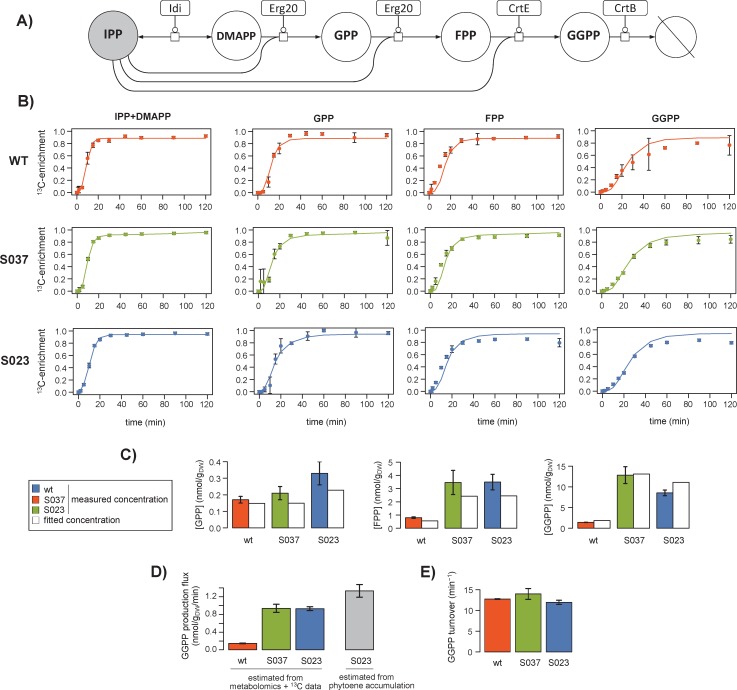
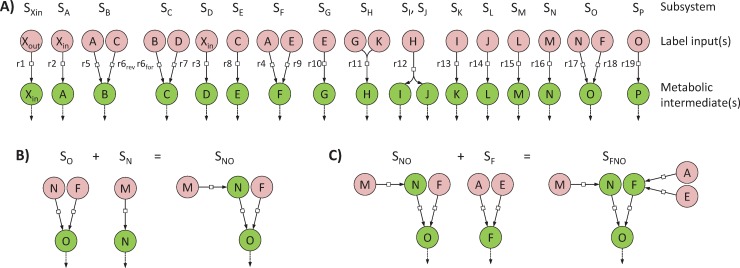
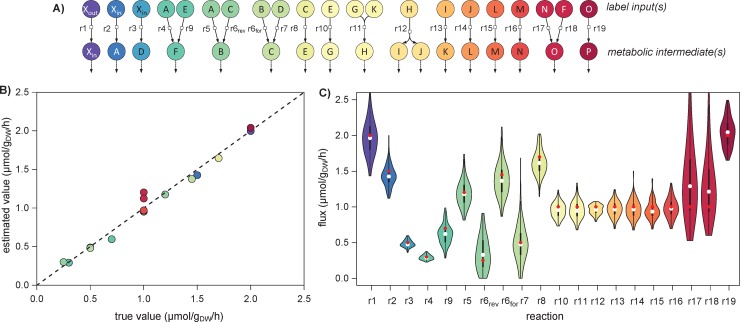
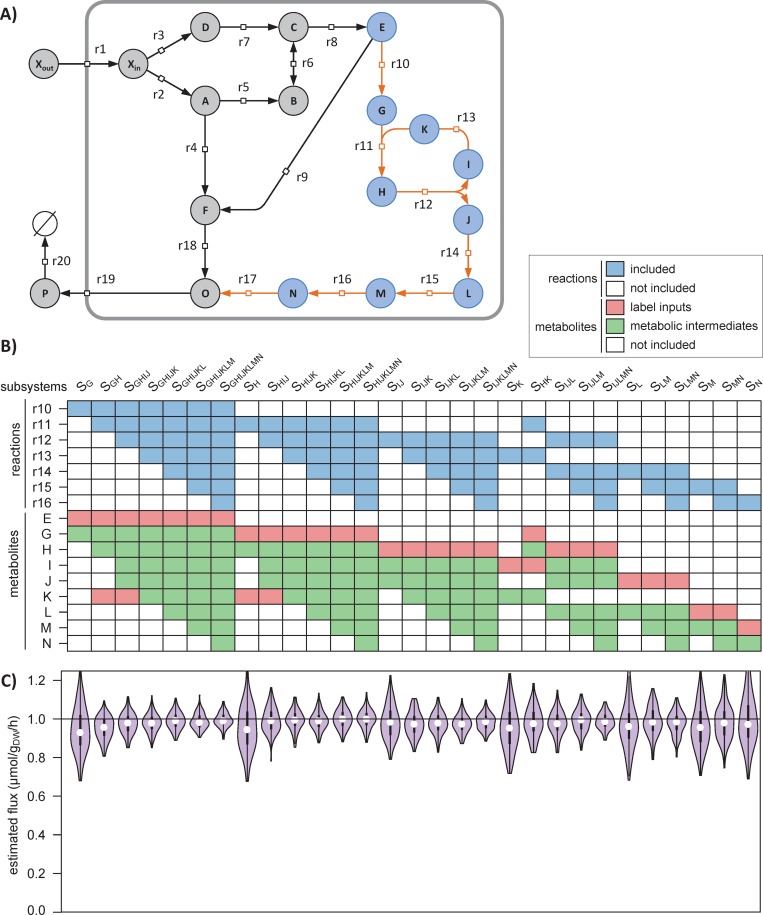
13C-metabolic flux analysis (13C-MFA) allows metabolic fluxes to be quantified in living organisms and is a major tool in biotechnology and systems biology. Current 13C-MFA approaches model label propagation starting from the extracellular 13C-labeled nutrient(s), which limits their applicability to the analysis of pathways close to this metabolic entry point. Here, we propose a new approach to quantify fluxes through any metabolic subnetwork of interest by modeling label propagation directly from the metabolic precursor(s) of this subnetwork. The flux calculations are thus purely based on information from within the subnetwork of interest, and no additional knowledge about the surrounding network (such as atom transitions in upstream reactions or the labeling of the extracellular nutrient) is required. This approach, termed ScalaFlux for SCALAble metabolic FLUX analysis, can be scaled up from individual reactions to pathways to sets of pathways. ScalaFlux has several benefits compared with current 13C-MFA approaches: greater network coverage, lower data requirements, independence from cell physiology, robustness to gaps in data and network information, better computational efficiency, applicability to rich media, and enhanced flux identifiability. We validated ScalaFlux using a theoretical network and simulated data. We also used the approach to quantify fluxes through the prenyl pyrophosphate pathway of Saccharomyces cerevisiae mutants engineered to produce phytoene, using a dataset for which fluxes could not be calculated using existing approaches. A broad range of metabolic systems can be targeted with minimal cost and effort, making ScalaFlux a valuable tool for the analysis of metabolic fluxes.
13C 代谢通量分析(13C-MFA)可定量测定活生物体中的代谢通量,是生物技术和系统生物学的主要工具。目前的 13C-MFA 方法从细胞外 13C 标记的营养物开始对标记传播进行建模,这限制了它们在接近该代谢入口途径的分析中的适用性。在这里,我们提出了一种新方法,通过直接从该亚网络的代谢前体来建模标记传播,从而对任何感兴趣的代谢子网络进行通量定量。因此,通量计算纯粹基于感兴趣的子网内的信息,不需要关于周围网络的任何其他知识(例如上游反应中的原子跃迁或细胞外营养物的标记)。这种方法称为 ScalaFlux(用于可扩展代谢通量分析的 SCALAble),可以从小分子反应扩展到途径再扩展到途径集。与当前的 13C-MFA 方法相比,ScalaFlux 具有几个优势:更大的网络覆盖范围、更低的数据要求、不依赖细胞生理学、对数据和网络信息中的空白具有鲁棒性、更好的计算效率、适用于丰富的培养基以及增强的通量可识别性。我们使用理论网络和模拟数据验证了 ScalaFlux。我们还使用该方法来量化酿酒酵母突变体中类异戊二烯焦磷酸途径的通量,这些突变体经过工程改造以产生番茄红素,使用现有的方法无法计算这些突变体的通量。可以以最小的成本和精力针对广泛的代谢系统进行靶向处理,这使得 ScalaFlux 成为代谢通量分析的有价值的工具。