Computational Biology Institute, Milken Institute School of Public Health, The George Washington University, Washington, DC 20052, USA.
Department of Biostatistics and Bioinformatics, Milken Institute School of Public Health, The George Washington University, Washington, DC 20052, USA.
Viruses. 2020 May 19;12(5):560. doi: 10.3390/v12050560.
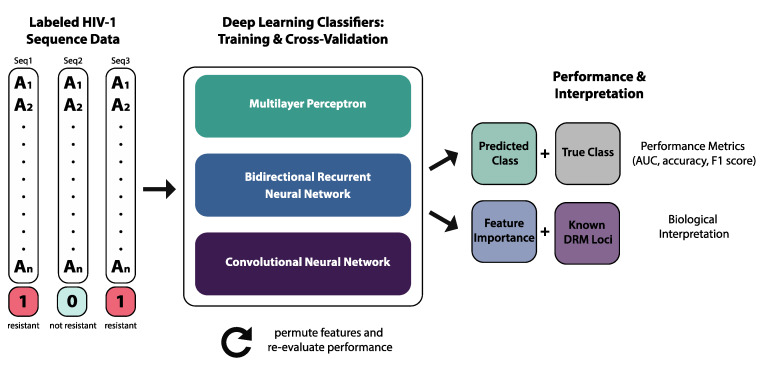
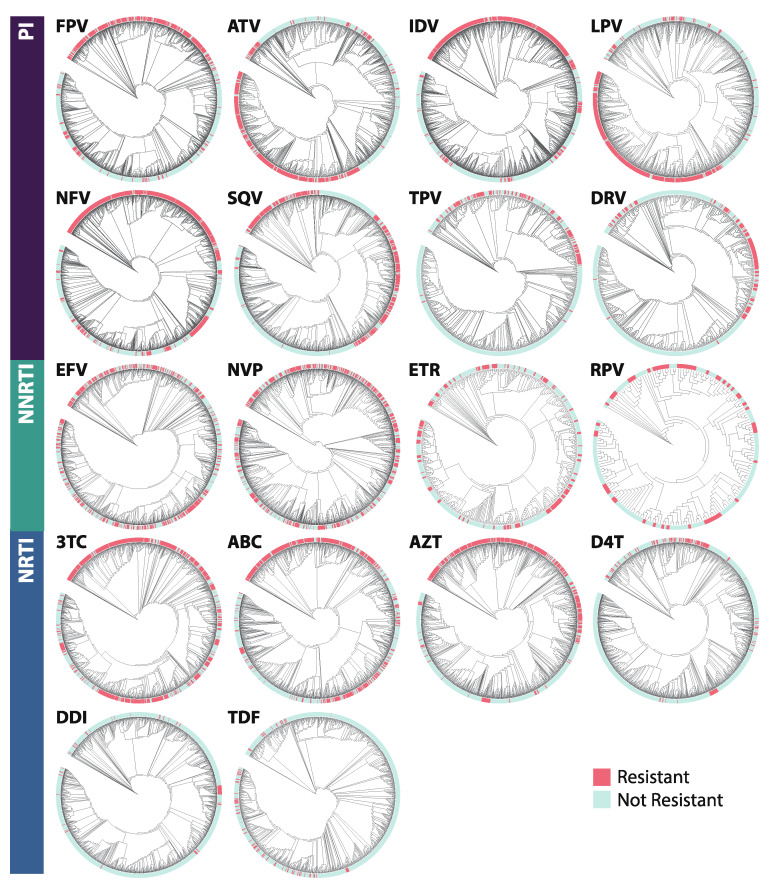
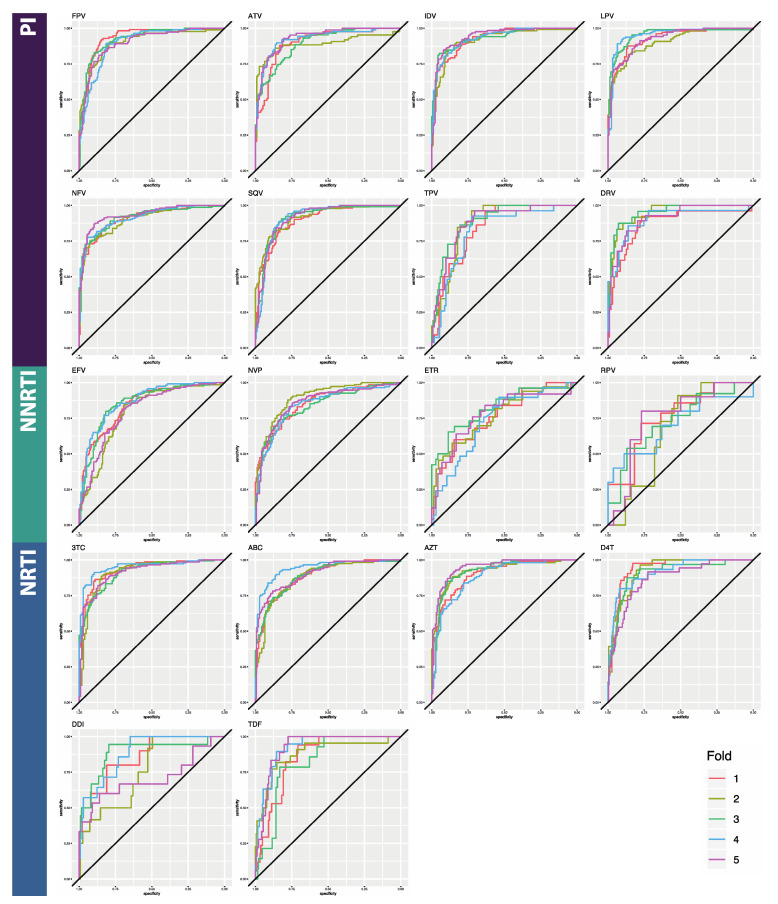
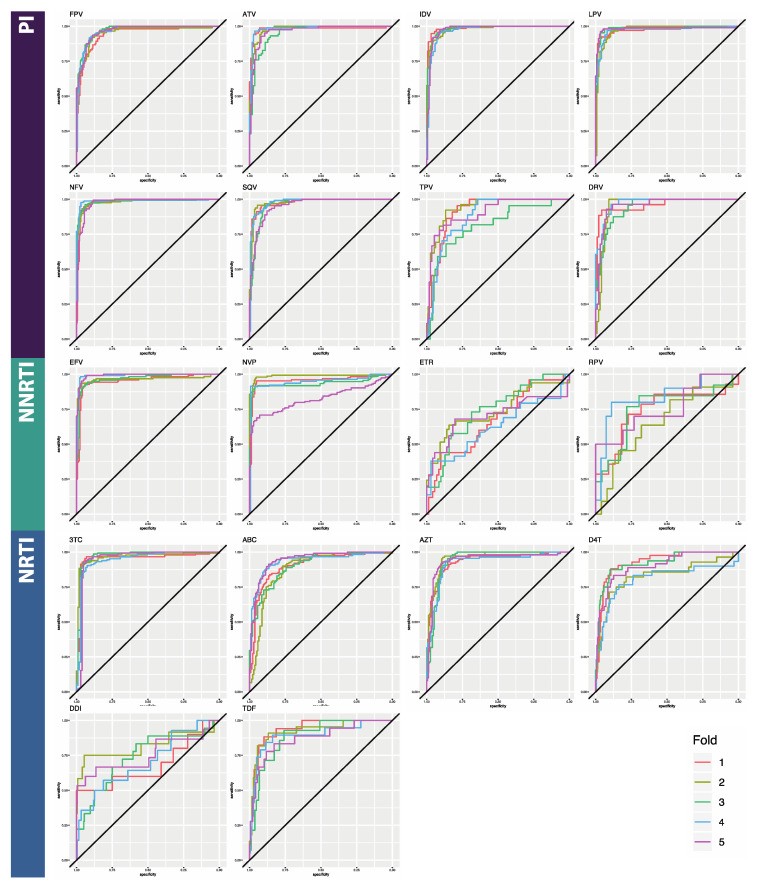
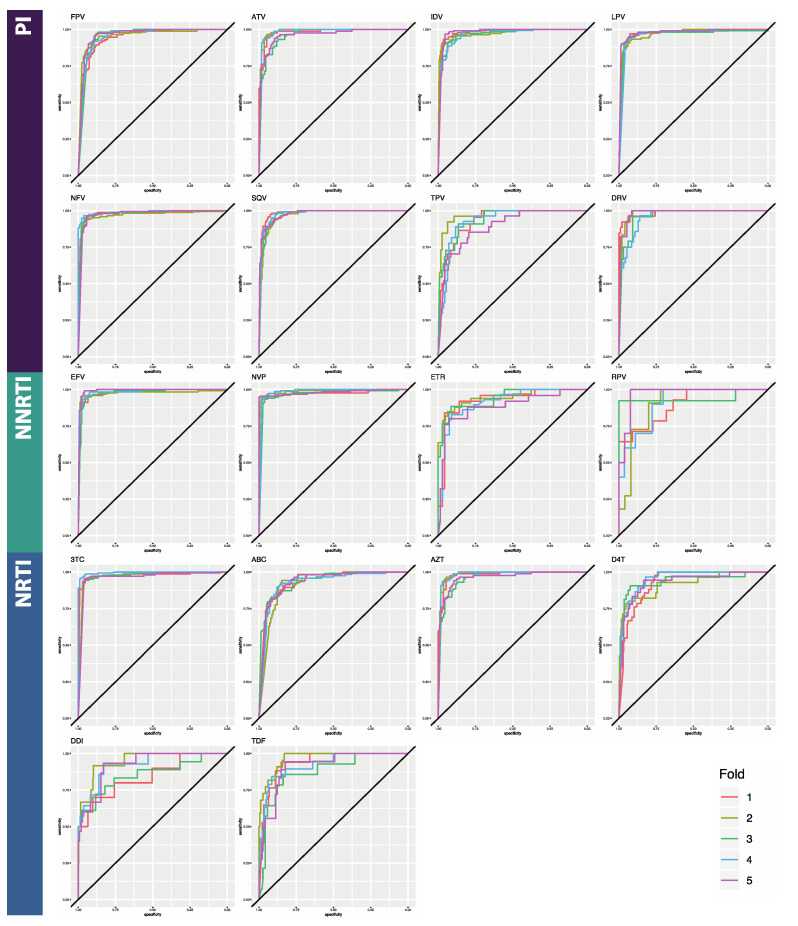
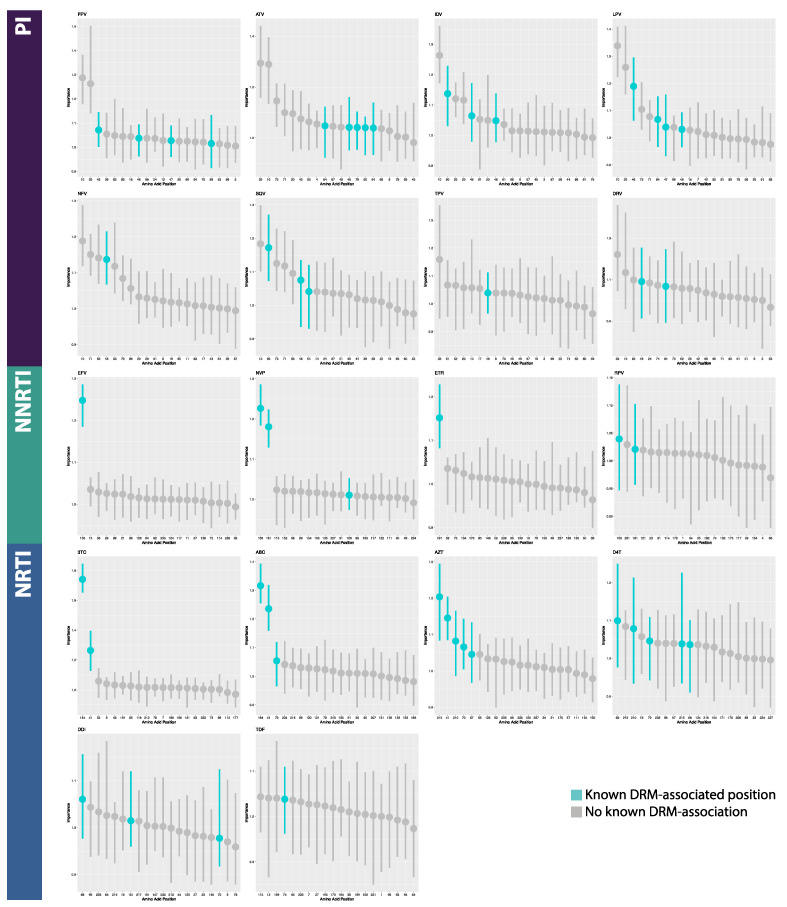
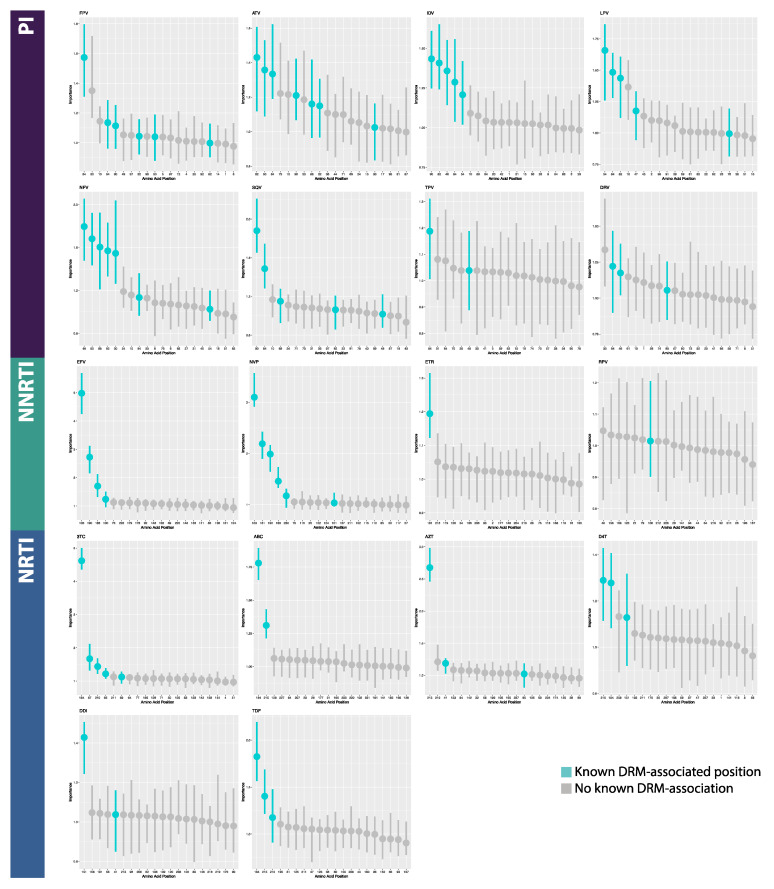
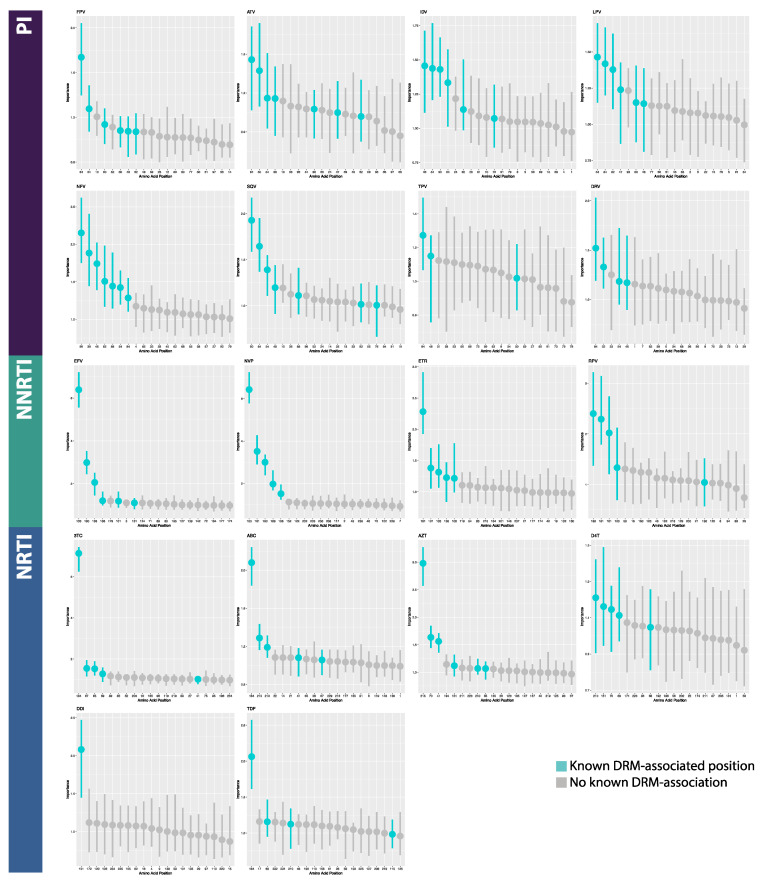
The fast replication rate and lack of repair mechanisms of human immunodeficiency virus (HIV) contribute to its high mutation frequency, with some mutations resulting in the evolution of resistance to antiretroviral therapies (ART). As such, studying HIV drug resistance allows for real-time evaluation of evolutionary mechanisms. Characterizing the biological process of drug resistance is also critically important for sustained effectiveness of ART. Investigating the link between "black box" deep learning methods applied to this problem and evolutionary principles governing drug resistance has been overlooked to date. Here, we utilized publicly available HIV-1 sequence data and drug resistance assay results for 18 ART drugs to evaluate the performance of three architectures (multilayer perceptron, bidirectional recurrent neural network, and convolutional neural network) for drug resistance prediction, jointly with biological analysis. We identified convolutional neural networks as the best performing architecture and displayed a correspondence between the importance of biologically relevant features in the classifier and overall performance. Our results suggest that the high classification performance of deep learning models is indeed dependent on drug resistance mutations (DRMs). These models heavily weighted several features that are not known DRM locations, indicating the utility of model interpretability to address causal relationships in viral genotype-phenotype data.
人类免疫缺陷病毒 (HIV) 的快速复制率和缺乏修复机制导致其突变频率很高,其中一些突变导致对抗逆转录病毒疗法 (ART) 的耐药性进化。因此,研究 HIV 耐药性可以实时评估进化机制。描述耐药性的生物学过程对于 ART 的持续有效性也至关重要。迄今为止,人们忽视了将应用于该问题的“黑盒”深度学习方法与控制耐药性的进化原则之间的联系进行研究。在这里,我们利用公开的 HIV-1 序列数据和 18 种 ART 药物的耐药性测定结果,评估了三种架构(多层感知器、双向递归神经网络和卷积神经网络)在耐药性预测方面的性能,并进行了联合生物学分析。我们发现卷积神经网络是表现最佳的架构,并显示出分类器中生物学相关特征的重要性与整体性能之间存在对应关系。我们的结果表明,深度学习模型的高分类性能确实取决于耐药性突变 (DRMs)。这些模型严重依赖于几个并非已知 DRM 位置的特征,这表明模型可解释性可用于解决病毒基因型-表型数据中的因果关系。