Biotechnology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran.
School of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran.
Sci Rep. 2020 Jul 2;10(1):10816. doi: 10.1038/s41598-020-67643-w.
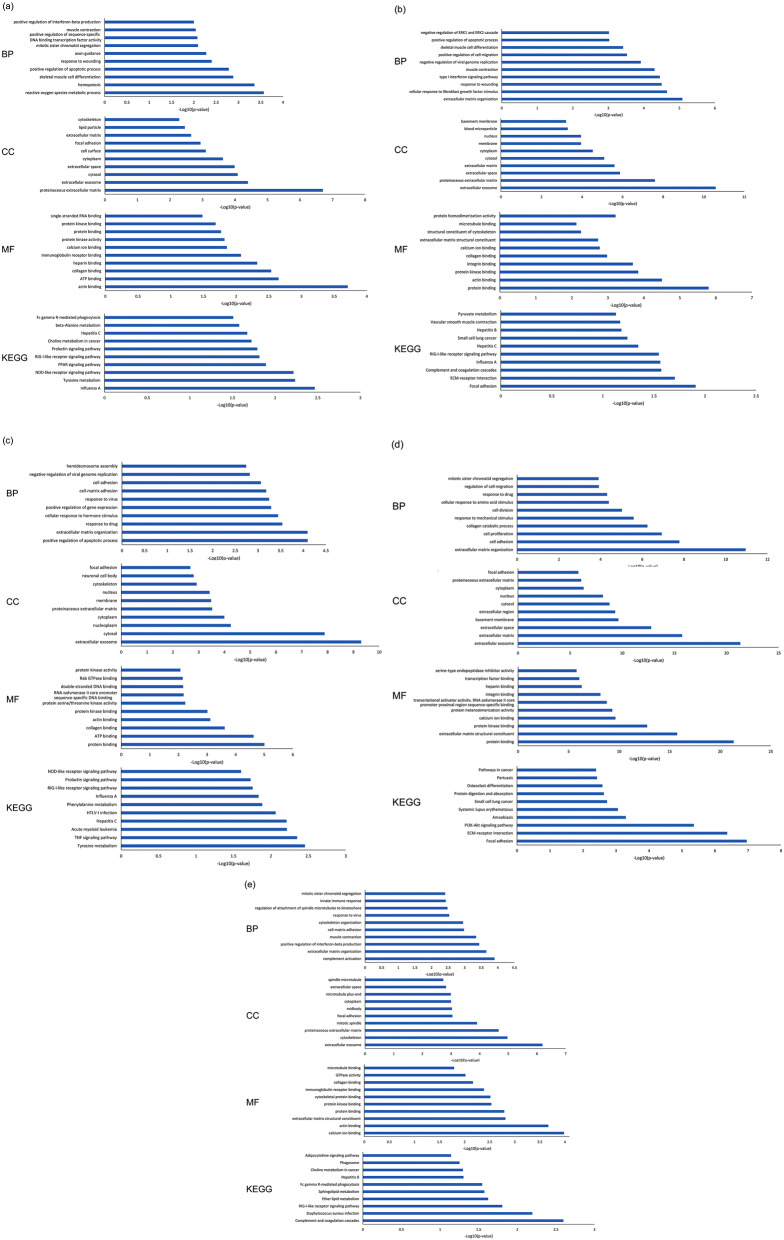
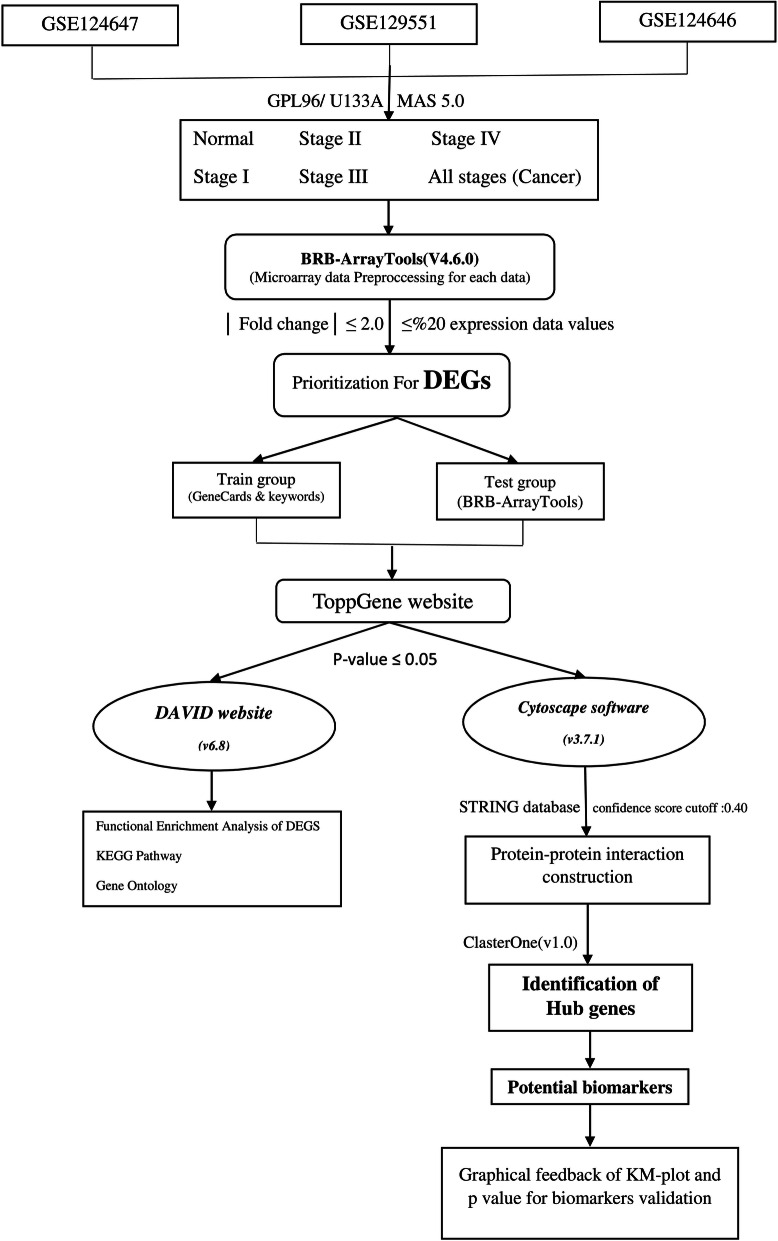
Breast cancer (BC), as one of the leading causes of death among women, comprises several subtypes with controversial and poor prognosis. Considering the TNM (tumor, lymph node, metastasis) based classification for staging of breast cancer, it is essential to diagnose the disease at early stages. The present study aims to take advantage of the systems biology approach on genome wide gene expression profiling datasets to identify the potential biomarkers involved at stage I, stage II, stage III, and stage IV as well as in the integrated group. Three HER2-negative breast cancer microarray datasets were retrieved from the GEO database, including normal, stage I, stage II, stage III, and stage IV samples. Additionally, one dataset was also extracted to test the developed predictive models trained on the three datasets. The analysis of gene expression profiles to identify differentially expressed genes (DEGs) was performed after preprocessing and normalization of data. Then, statistically significant prioritized DEGs were used to construct protein-protein interaction networks for the stages for module analysis and biomarker identification. Furthermore, the prioritized DEGs were used to determine the involved GO enrichment and KEGG signaling pathways at various stages of the breast cancer. The recurrence survival rate analysis of the identified gene biomarkers was conducted based on Kaplan-Meier methodology. Furthermore, the identified genes were validated not only by using several classification models but also through screening the experimental literature reports on the target genes. Fourteen (21 genes), nine (17 genes), eight (10 genes), four (7 genes), and six (8 genes) gene modules (total of 53 unique genes out of 63 genes with involving those with the same connectivity degree) were identified for stage I, stage II, stage III, stage IV, and the integrated group. Moreover, SMC4, FN1, FOS, JUN, and KIF11 and RACGAP1 genes with the highest connectivity degrees were in module 1 for abovementioned stages, respectively. The biological processes, cellular components, and molecular functions were demonstrated for outcomes of GO analysis and KEGG pathway assessment. Additionally, the Kaplan-Meier analysis revealed that 33 genes were found to be significant while considering the recurrence-free survival rate as an alternative to overall survival rate. Furthermore, the machine learning calcification models show good performance on the determined biomarkers. Moreover, the literature reports have confirmed all of the identified gene biomarkers for breast cancer. According to the literature evidence, the identified hub genes are highly correlated with HER2-negative breast cancer. The 53-mRNA signature might be a potential gene set for TNM based stages as well as possible therapeutics with potentially good performance in predicting and managing recurrence-free survival rates at stages I, II, III, and IV as well as in the integrated group. Moreover, the identified genes for the TNM-based stages can also be used as mRNA profile signatures to determine the current stage of the breast cancer.
乳腺癌(BC)是女性死亡的主要原因之一,包括几种具有争议和预后不良的亚型。考虑到基于肿瘤(tumor)、淋巴结(lymph node)、转移(metastasis)的 TNM 分类用于乳腺癌分期,早期诊断疾病至关重要。本研究旨在利用系统生物学方法对全基因组基因表达谱数据集进行分析,以确定 I 期、II 期、III 期和 IV 期以及整合组中涉及的潜在生物标志物。从 GEO 数据库中检索了三个 HER2 阴性乳腺癌微阵列数据集,包括正常、I 期、II 期、III 期和 IV 期样本。此外,还提取了一个数据集来测试在三个数据集上训练的开发预测模型。在对数据进行预处理和归一化后,对基因表达谱进行分析以识别差异表达基因(DEGs)。然后,使用具有统计学意义的优先 DEGs 构建用于各阶段的蛋白质-蛋白质相互作用网络,以进行模块分析和生物标志物识别。此外,还使用优先 DEGs 确定乳腺癌各阶段涉及的 GO 富集和 KEGG 信号通路。基于 Kaplan-Meier 方法对鉴定的基因生物标志物的复发生存率进行分析。此外,不仅通过使用几种分类模型,还通过筛选目标基因的实验文献报告,对鉴定的基因进行了验证。为 I 期、II 期、III 期、IV 期和整合组分别鉴定了 14 个(21 个基因)、9 个(17 个基因)、8 个(10 个基因)、4 个(7 个基因)和 6 个(8 个基因)基因模块(63 个具有相同连接度的基因中共有 53 个独特基因)。此外,在上述各个阶段的模块 1 中,具有最高连接度的基因 SMC4、FN1、FOS、JUN 和 KIF11 以及 RACGAP1 分别为 SMC4、FN1、FOS、JUN 和 KIF11 以及 RACGAP1。GO 分析和 KEGG 途径评估的结果表明了生物学过程、细胞成分和分子功能。此外,Kaplan-Meier 分析显示,考虑到无复发生存率作为总生存率的替代指标,有 33 个基因是显著的。此外,机器学习钙化模型在确定的生物标志物上表现出良好的性能。此外,文献报道已经证实了所有鉴定的乳腺癌基因生物标志物。根据文献证据,鉴定的枢纽基因与 HER2 阴性乳腺癌高度相关。53-mRNA 特征可能是基于 TNM 的阶段的潜在基因集,以及在预测和管理 I 期、II 期、III 期和 IV 期以及整合组中的无复发生存率方面具有潜在良好性能的潜在治疗方法。此外,用于基于 TNM 的阶段的鉴定基因也可作为 mRNA 谱特征用于确定乳腺癌的当前阶段。