Richmond Phillip A, van der Kloet Frans, Vaz Frederic M, Lin David, Uzozie Anuli, Graham Emma, Kobor Michael, Mostafavi Sara, Moerland Perry D, Lange Philipp F, van Kampen Antoine H C, Wasserman Wyeth W, Engelen Marc, Kemp Stephan, van Karnebeek Clara D M
Center for Molecular Medicine and Therapeutics, BC Children's Hospital Research Institute, University of British Columbia, Vancouver, BC, Canada.
Bioinformatics Laboratory, Department of Clinical Epidemiology, Biostatistics and Bioinformatics, Amsterdam Public Health Research Institute, Amsterdam University Medical Center, University of Amsterdam, Amsterdam, Netherlands.
Front Cell Dev Biol. 2020 Jun 25;8:520. doi: 10.3389/fcell.2020.00520. eCollection 2020.
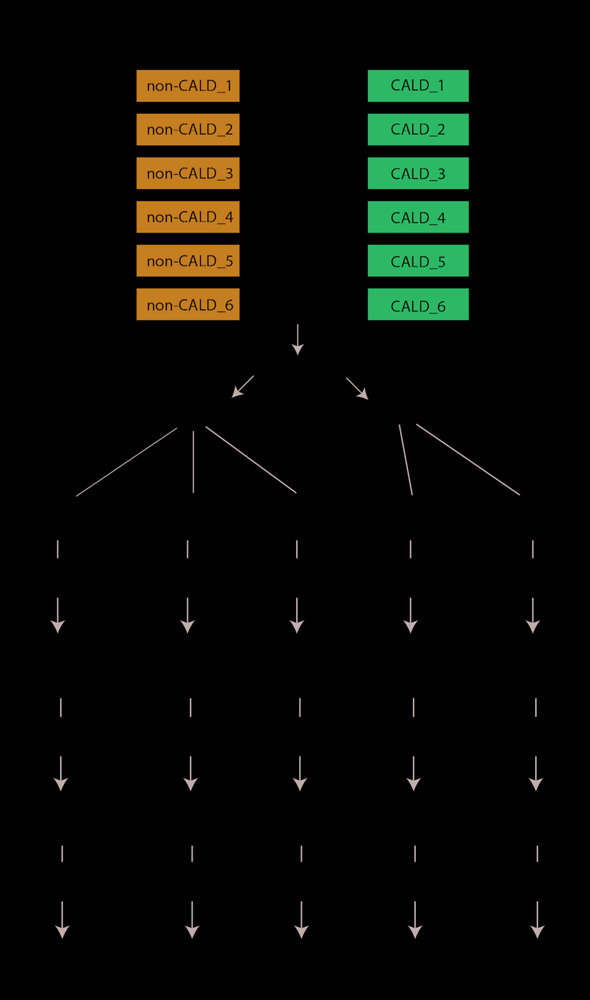
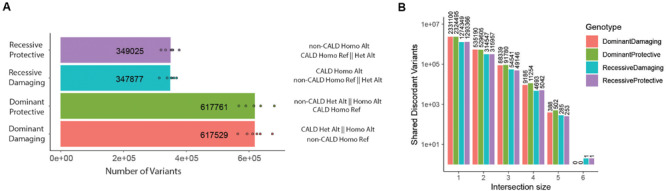
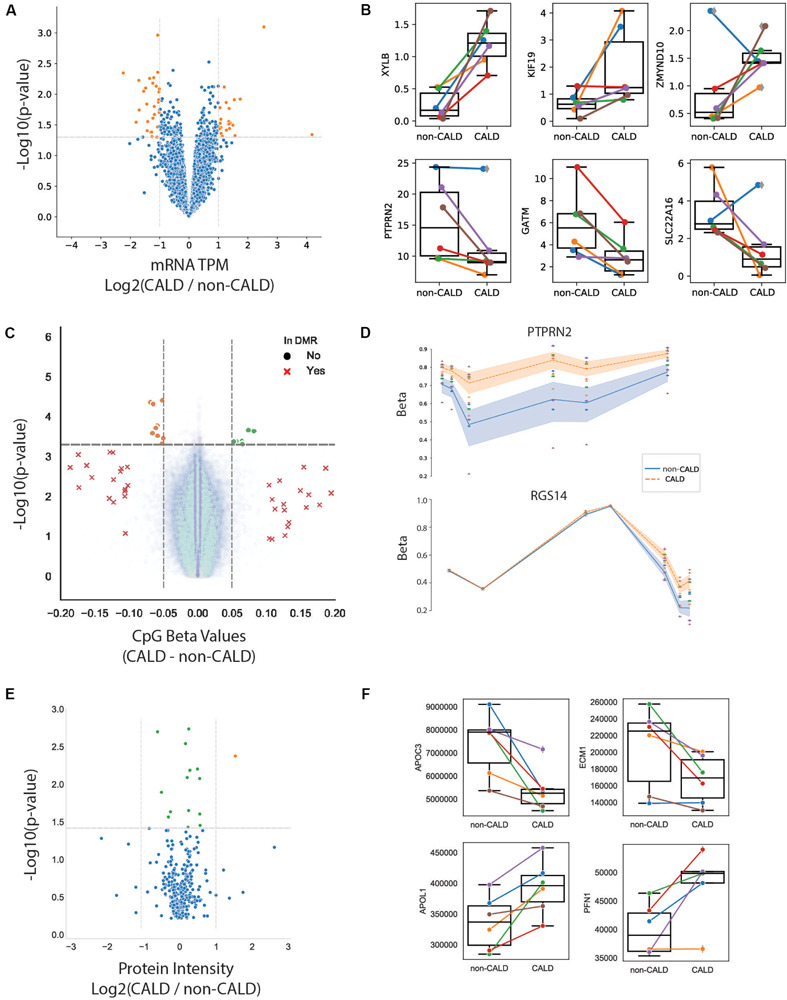
X-linked adrenoleukodystrophy (ALD) is a peroxisomal metabolic disorder with a highly complex clinical presentation. ALD is caused by mutations in the gene, and is characterized by the accumulation of very long-chain fatty acids in plasma and tissues. Disease-causing mutations are 'loss of function' mutations, with no prognostic value with respect to the clinical outcome of an individual. All male patients with ALD develop spinal cord disease and a peripheral neuropathy in adulthood, although age of onset is highly variable. However, the lifetime prevalence to develop progressive white matter lesions, termed cerebral ALD (CALD), is only about 60%. Early identification of transition to CALD is critical since it can be halted by allogeneic hematopoietic stem cell therapy only in an early stage. The primary goal of this study is to identify molecular markers which may be prognostic of cerebral demyelination from a simple blood sample, with the hope that blood-based assays can replace the current protocols for diagnosis. We collected six well-characterized brother pairs affected by ALD and discordant for the presence of CALD and performed multi-omic profiling of blood samples including genome, epigenome, transcriptome, metabolome/lipidome, and proteome profiling. In our analysis we identify discordant genomic alleles present across all families as well as differentially abundant molecular features across the omics technologies. The analysis was focused on univariate modeling to discriminate the two phenotypic groups, but was unable to identify statistically significant candidate molecular markers. Our study highlights the issues caused by a large amount of inter-individual variation, and supports the emerging hypothesis that cerebral demyelination is a complex mix of environmental factors and/or heterogeneous genomic alleles. We confirm previous observations about the role of immune response, specifically auto-immunity and the potential role of PFN1 protein overabundance in CALD in a subset of the families. We envision our methodology as well as dataset has utility to the field for reproducing previous or enabling future modifier investigations.
X连锁肾上腺脑白质营养不良(ALD)是一种过氧化物酶体代谢紊乱疾病,临床表现高度复杂。ALD由该基因的突变引起,其特征是血浆和组织中极长链脂肪酸的积累。致病突变是“功能丧失”突变,对个体的临床结果没有预后价值。所有男性ALD患者在成年后都会出现脊髓疾病和周围神经病变,尽管发病年龄差异很大。然而,发展为进行性白质病变(称为脑型ALD,CALD)的终生患病率仅约为60%。早期识别向CALD的转变至关重要,因为只有在早期阶段,同种异体造血干细胞疗法才能阻止其发展。本研究的主要目标是从简单的血液样本中识别可能对脑脱髓鞘具有预后作用的分子标记物,希望基于血液的检测方法能够取代当前的诊断方案。我们收集了六对特征明确的兄弟对,他们都患有ALD,但其中一方存在CALD,另一方不存在,并对血液样本进行了多组学分析,包括基因组、表观基因组、转录组、代谢组/脂质组和蛋白质组分析。在我们的分析中,我们识别出了所有家族中存在的不一致基因组等位基因以及跨组学技术差异丰富的分子特征。分析集中在单变量模型上,以区分两个表型组,但未能识别出具有统计学意义的候选分子标记物。我们的研究突出了个体间大量变异所导致的问题,并支持了新出现的假说,即脑脱髓鞘是环境因素和/或异质基因组等位基因的复杂组合。我们证实了先前关于免疫反应作用的观察结果,特别是自身免疫以及PFN1蛋白过量在部分家族的CALD中的潜在作用。我们设想我们的方法以及数据集对该领域具有实用性,可用于重现先前的研究或开展未来的修饰因子研究。