Department of Chemistry and Biochemistry; UCLA-DOE Institute for Genomics and Proteomics; STROBE, NSF Science and Technology Center, University of California Los Angeles (UCLA), Los Angeles, CA 90095, USA.
Crystallographic Methods, Institute of Molecular Biology of Barcelona (IBMB-CSIC), Barcelona Science Park, Helix Building, Baldiri Reixac 15, 08028 Barcelona, Spain.
Acta Crystallogr D Struct Biol. 2020 Aug 1;76(Pt 8):703-712. doi: 10.1107/S2059798320008049. Epub 2020 Jul 27.
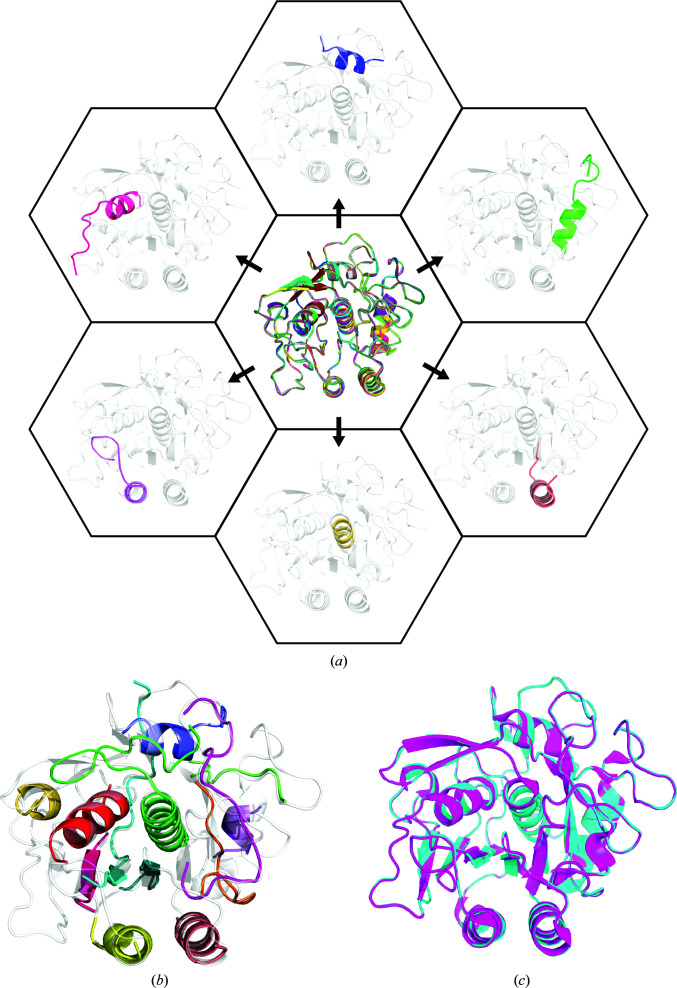
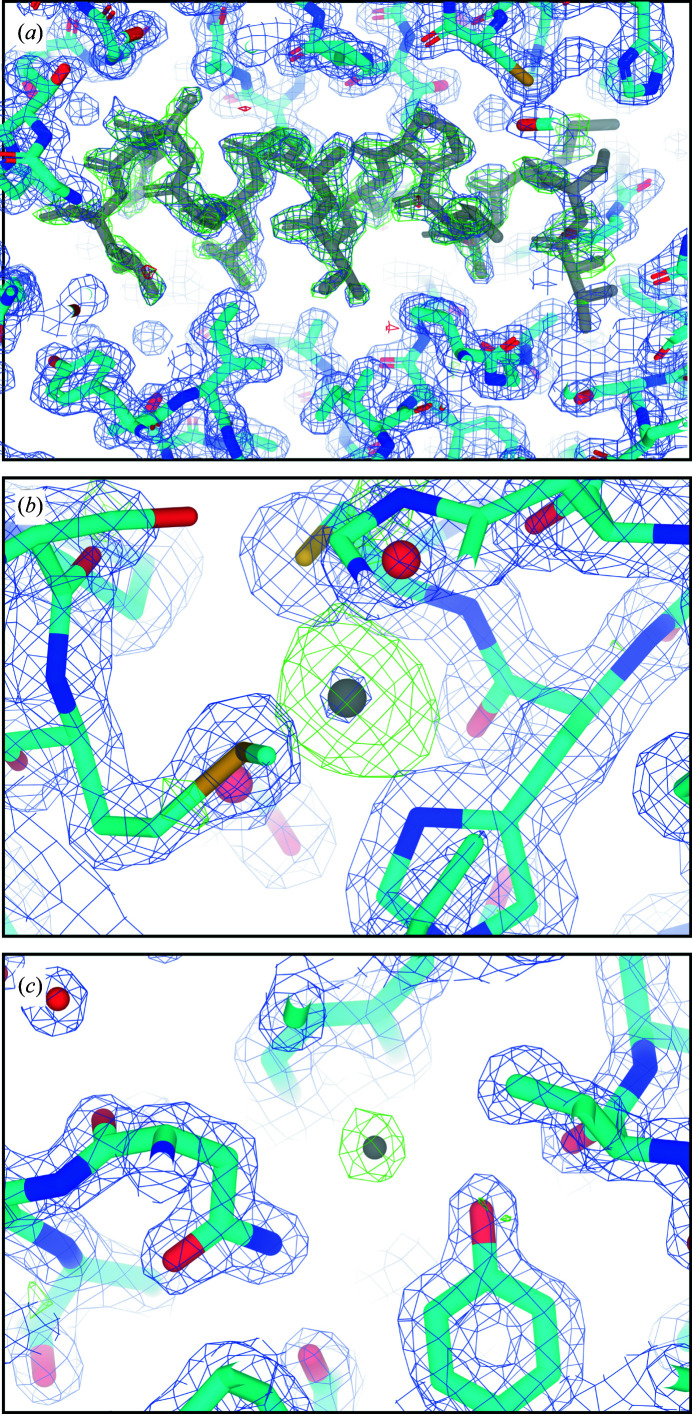
Structure determination of novel biological macromolecules by X-ray crystallography can be facilitated by the use of small structural fragments, some of only a few residues in length, as effective search models for molecular replacement to overcome the phase problem. Independence from the need for a complete pre-existing model with sequence similarity to the crystallized molecule is the primary appeal of ARCIMBOLDO, a suite of programs which employs this ab initio algorithm for phase determination. Here, the use of ARCIMBOLDO is investigated to overcome the phase problem with the electron cryomicroscopy (cryoEM) method known as microcrystal electron diffraction (MicroED). The results support the use of the ARCIMBOLDO_SHREDDER pipeline to provide phasing solutions for a structure of proteinase K from 1.6 Å resolution data using model fragments derived from the structures of proteins sharing a sequence identity of as low as 20%. ARCIMBOLDO_SHREDDER identified the most accurate polyalanine fragments from a set of distantly related sequence homologues. Alternatively, such templates were extracted in spherical volumes and given internal degrees of freedom to refine towards the target structure. Both modes relied on the rotation function in Phaser to identify or refine fragment models and its translation function to place them. Model completion from the placed fragments proceeded through phase combination of partial solutions and/or density modification and main-chain autotracing using SHELXE. The combined set of fragments was sufficient to arrive at a solution that resembled that determined by conventional molecular replacement using the known target structure as a search model. This approach obviates the need for a single, complete and highly accurate search model when phasing MicroED data, and permits the evaluation of large fragment libraries for this purpose.
通过 X 射线晶体学确定新型生物大分子的结构,可以使用小的结构片段作为有效搜索模型,进行分子置换,以克服相位问题。ARCIMBOLDO 是一套程序,它采用这种从头算法进行相位测定,不依赖于与结晶分子具有序列相似性的完整预先存在的模型,这是其主要吸引力。在这里,研究了使用 ARCIMBOLDO 来克服电子晶体显微镜(cryoEM)方法(称为微晶电子衍射(MicroED))的相位问题。结果支持使用 ARCIMBOLDO_SHREDDER 流水线为蛋白酶 K 的结构提供相位解决方案,该结构的分辨率为 1.6 Å,使用源自共享序列同一性低至 20%的蛋白质结构的模型片段。ARCIMBOLDO_SHREDDER 从一组远缘序列同源物中确定了最准确的多聚丙氨酸片段。或者,以球形体积提取这些模板,并赋予其内部自由度,以使其向目标结构细化。这两种模式都依赖于 Phaser 中的旋转函数来识别或细化片段模型,以及其平移函数来放置它们。通过使用 SHELXE 对部分解决方案进行相位组合和/或密度修饰以及主链自动跟踪,从放置的片段进行模型完成。放置片段的组合足以得到类似于使用已知目标结构作为搜索模型进行常规分子置换确定的解决方案。这种方法避免了在相位 MicroED 数据时需要单个完整且高度准确的搜索模型,并允许为此目的评估大型片段库。