The Third Clinical Medical College of Zhejiang Chinese Medical University, Hangzhou, Zhejiang 310051, China.
The First Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou, Zhejiang 310002, China.
Biomed Res Int. 2020 Aug 3;2020:6943103. doi: 10.1155/2020/6943103. eCollection 2020.
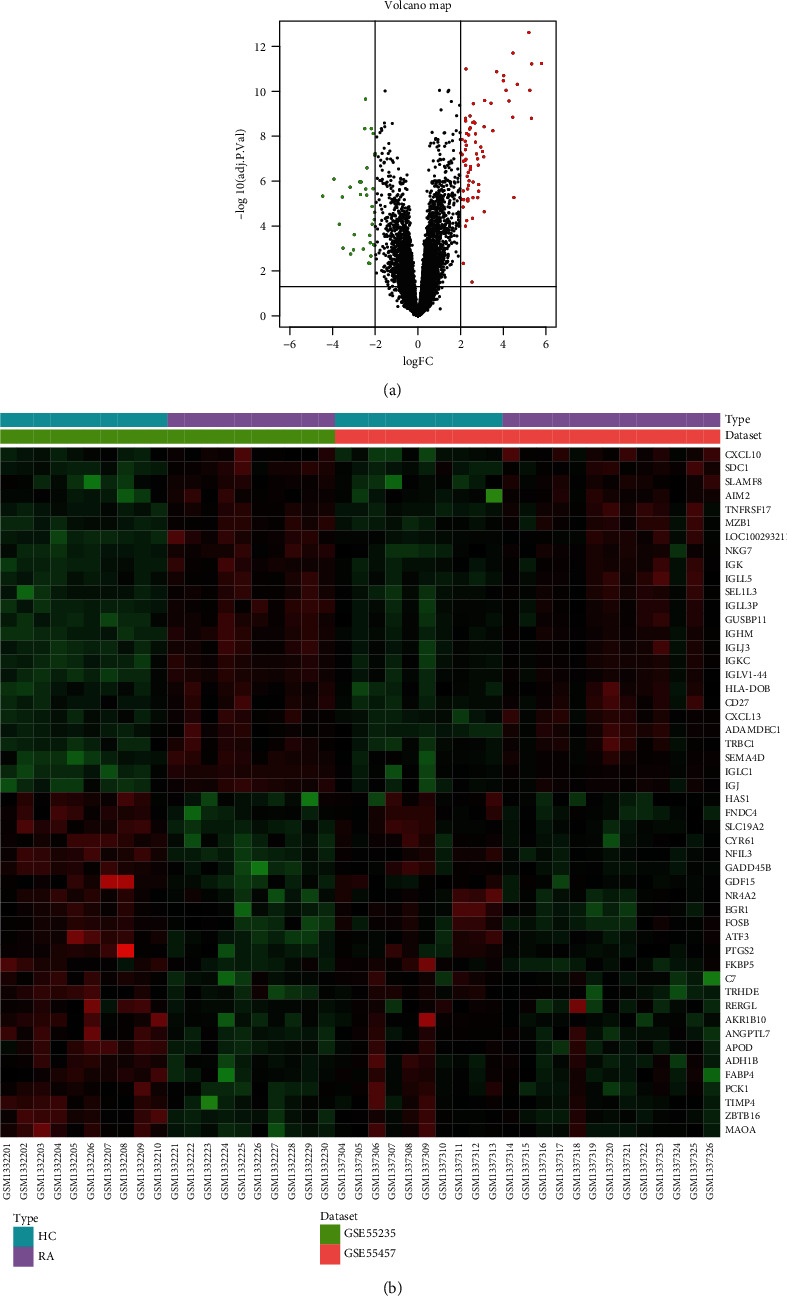
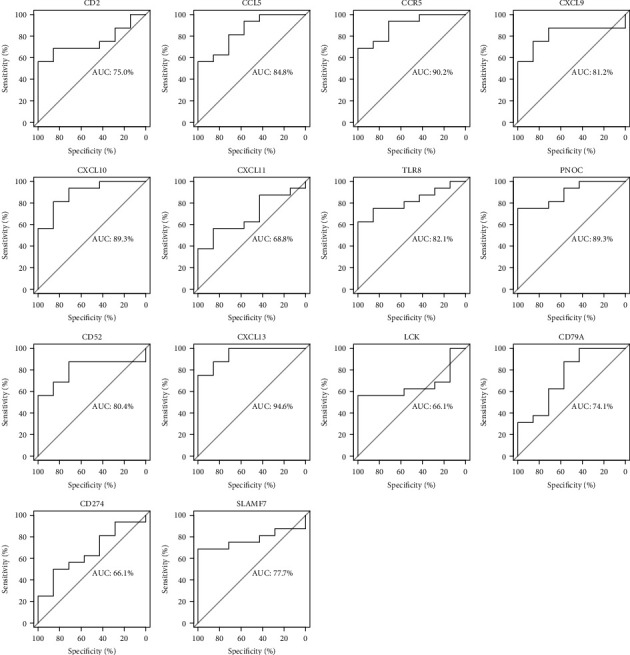
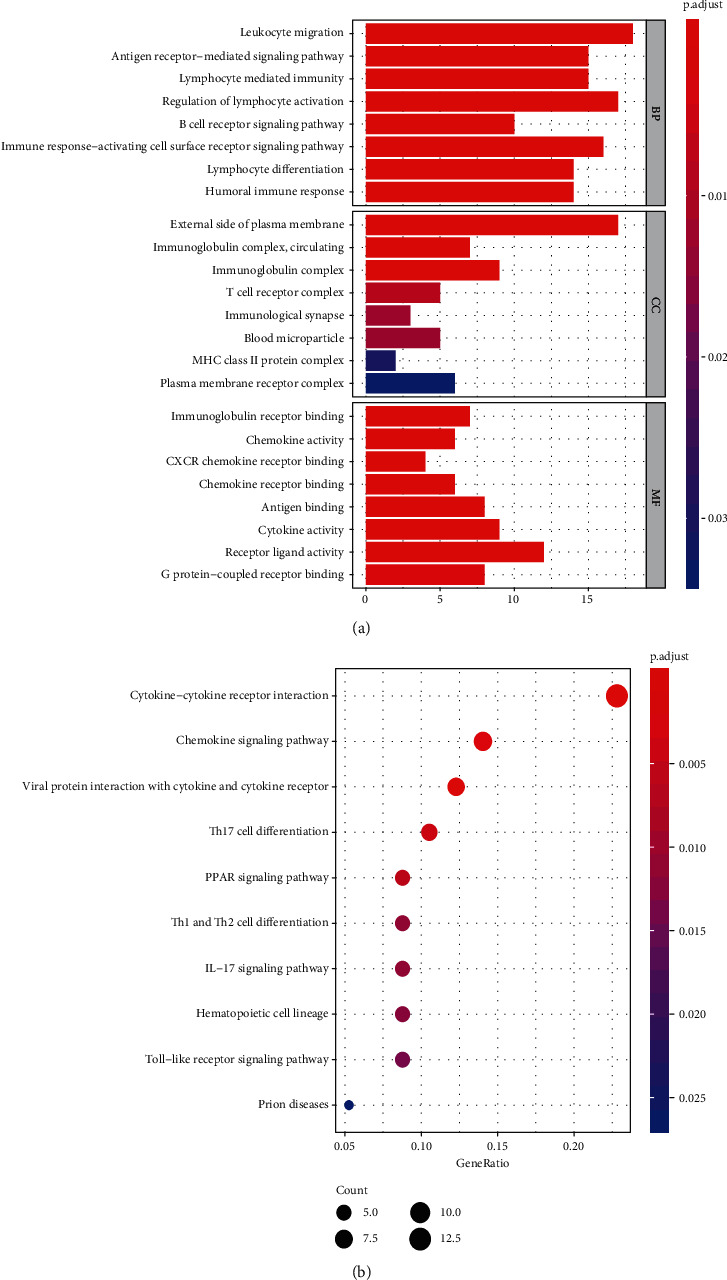
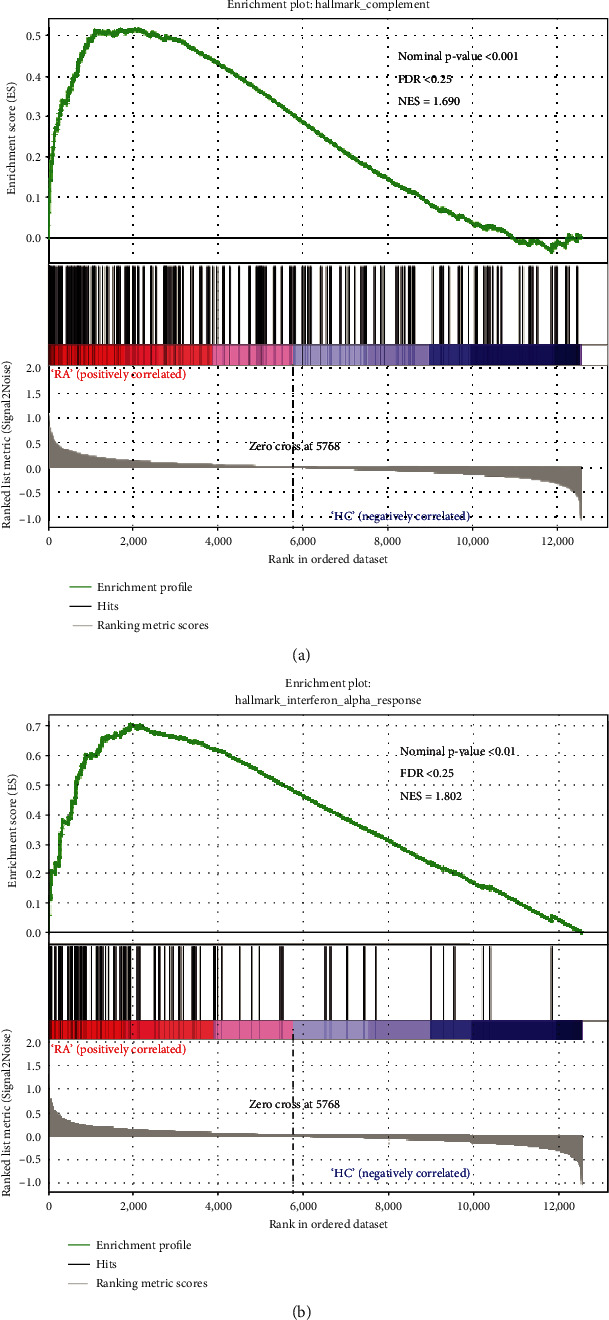
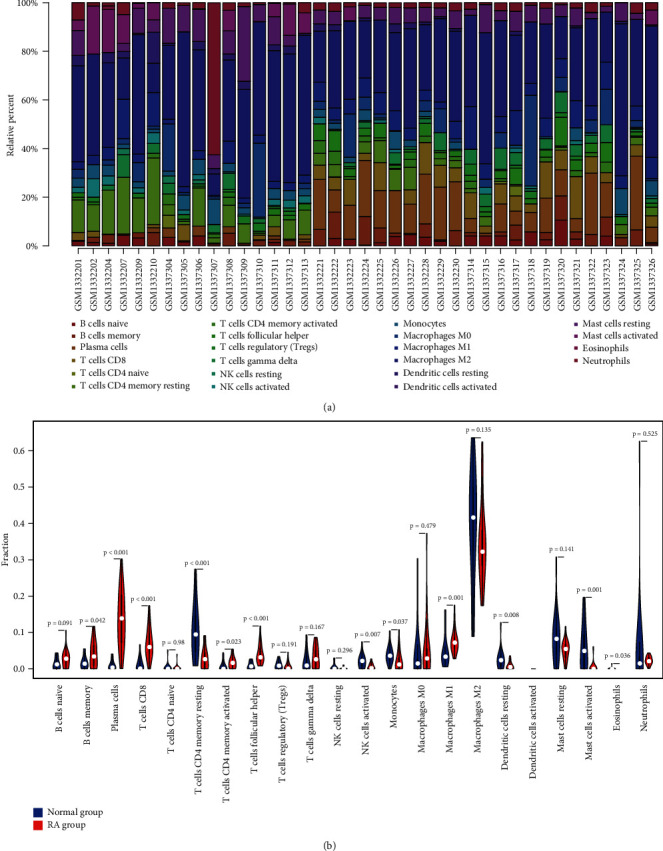
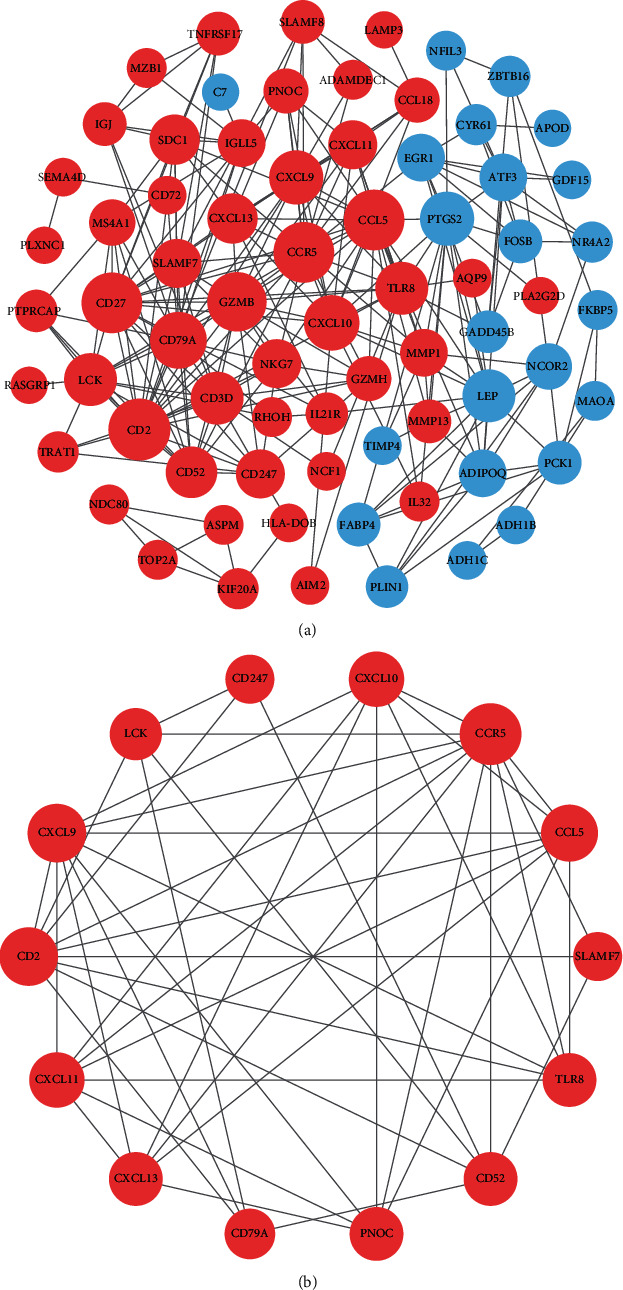
Rheumatoid arthritis (RA) is an autoimmune disease characterized by erosive arthritis, which has not been thoroughly cured yet, and standardized treatment is helpful for alleviating clinical symptoms. Here, various bioinformatics analysis tools were comprehensively utilized, aiming to identify critical biomarkers and possible pathogenesis of RA. Three gene expression datasets profiled by microarray were obtained from GEO database. Dataset GSE55235 and GSE55457 were merged for subsequent analyses. We identified differentially expressed genes (DEGs) in RStudio with limma package, performing functional enrichment analysis based on GSEA software and clusterProfiler package. Next, protein-protein interaction (PPI) network was set up through STRING database and Cytoscape. Moreover, CIBERSORT website was used to assess the inflammatory state of RA. Finally, we validated the candidate hub genes with dataset GSE77298. As a result, we identified 106 DEGs (72 upregulated and 34 downregulated genes). Through GO, KEGG, and GSEA analysis, we found that DEGs were mainly involved in immune response and inflammatory signaling pathway. With the help of Cytoscape software and MCODE plug-in, the most prominent subnetwork was screened out, containing 14 genes and 45 edges. For ROC curve analysis, eight genes with AUC >0.80 were considered as hub genes of RA. In conclusion, compared with healthy controls, the DEGs and their closely related biological functions were analyzed, and we held that chemokines and immune cells infiltration promote the progression of rheumatoid arthritis. Targeting the eight biomarkers we identified may be useful for the diagnosis and treatment of rheumatoid arthritis.
类风湿关节炎(RA)是一种自身免疫性疾病,其特征为侵蚀性关节炎,目前尚未彻底治愈,标准化治疗有助于缓解临床症状。本研究综合利用多种生物信息学分析工具,旨在鉴定 RA 的关键生物标志物和可能的发病机制。从 GEO 数据库中获取了三个基于微阵列的基因表达数据集。在 RStudio 中使用 limma 包合并数据集 GSE55235 和 GSE55457,基于 GSEA 软件和 clusterProfiler 包进行功能富集分析。接下来,通过 STRING 数据库和 Cytoscape 构建蛋白质-蛋白质相互作用(PPI)网络。此外,使用 CIBERSORT 网站评估 RA 的炎症状态。最后,使用数据集 GSE77298 验证候选枢纽基因。结果共鉴定出 106 个 DEGs(72 个上调和 34 个下调基因)。通过 GO、KEGG 和 GSEA 分析发现,DEGs 主要参与免疫反应和炎症信号通路。借助 Cytoscape 软件和 MCODE 插件筛选出最显著的子网络,包含 14 个基因和 45 个边缘。通过 ROC 曲线分析,将 AUC>0.80 的 8 个基因视为 RA 的枢纽基因。综上所述,与健康对照组相比,分析了 DEGs 及其密切相关的生物学功能,认为趋化因子和免疫细胞浸润促进类风湿关节炎的进展。针对我们鉴定的八个生物标志物可能有助于类风湿关节炎的诊断和治疗。