Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, WI, USA.
Institute of Clinical Medicine Research, Department of Gastroenterology and Hepatology, Suzhou Hospital affiliated with Nanjing Medical University, Suzhou, Jiangsu, China.
Cell Physiol Biochem. 2020 Sep 9;54(5):853-874. doi: 10.33594/000000274.
BACKGROUND/AIMS: The role of VDAC1, the most abundant mitochondrial outer membrane protein, in cell death depends on cell types and stimuli. Both silencing and upregulation of VDAC1 in various type of cancer cell lines can stimulate apoptosis. In contrast, in mouse embryonic stem (MES) cells and mouse embryonic fibroblasts (MEFs), the roles of VDAC1 knockout (VDAC1) in apoptotic cell death are contradictory. The contribution and underlying mechanism of VDAC1 in oxidative stress-induced cell death in cardiac cells has not been established. We hypothesized that VDAC1 is an essential regulator of oxidative stress-induced cell death in H9c2 cells.
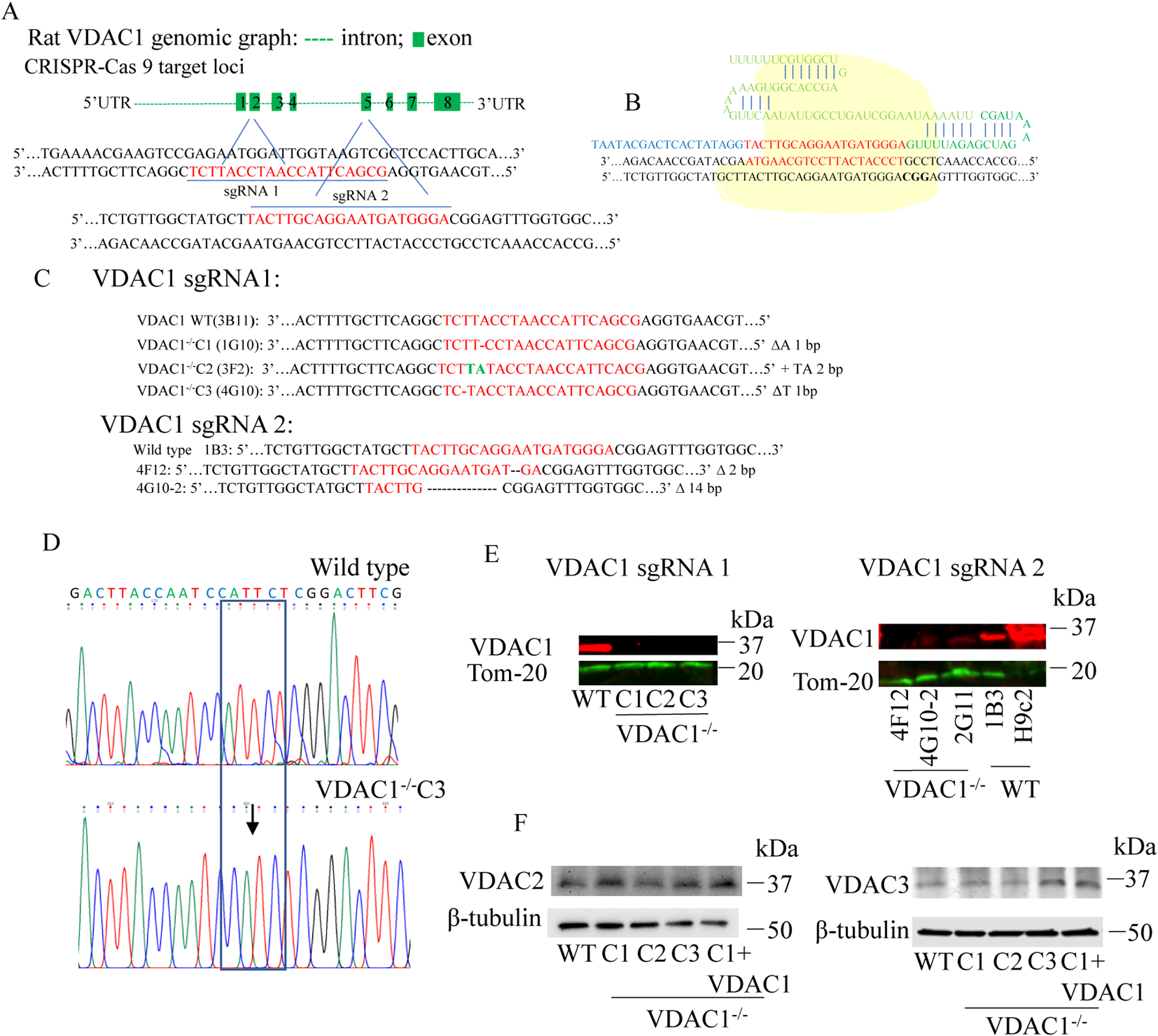
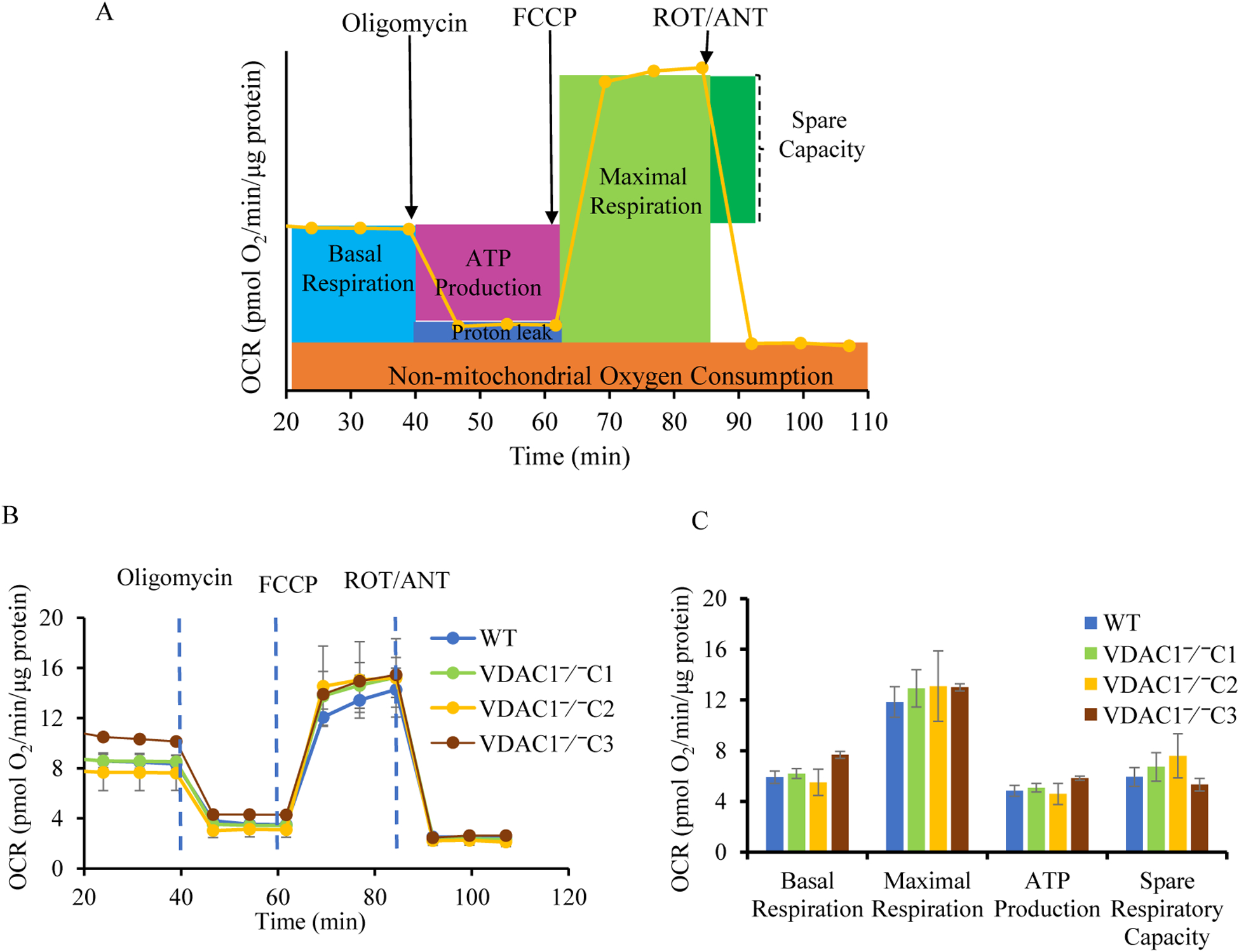
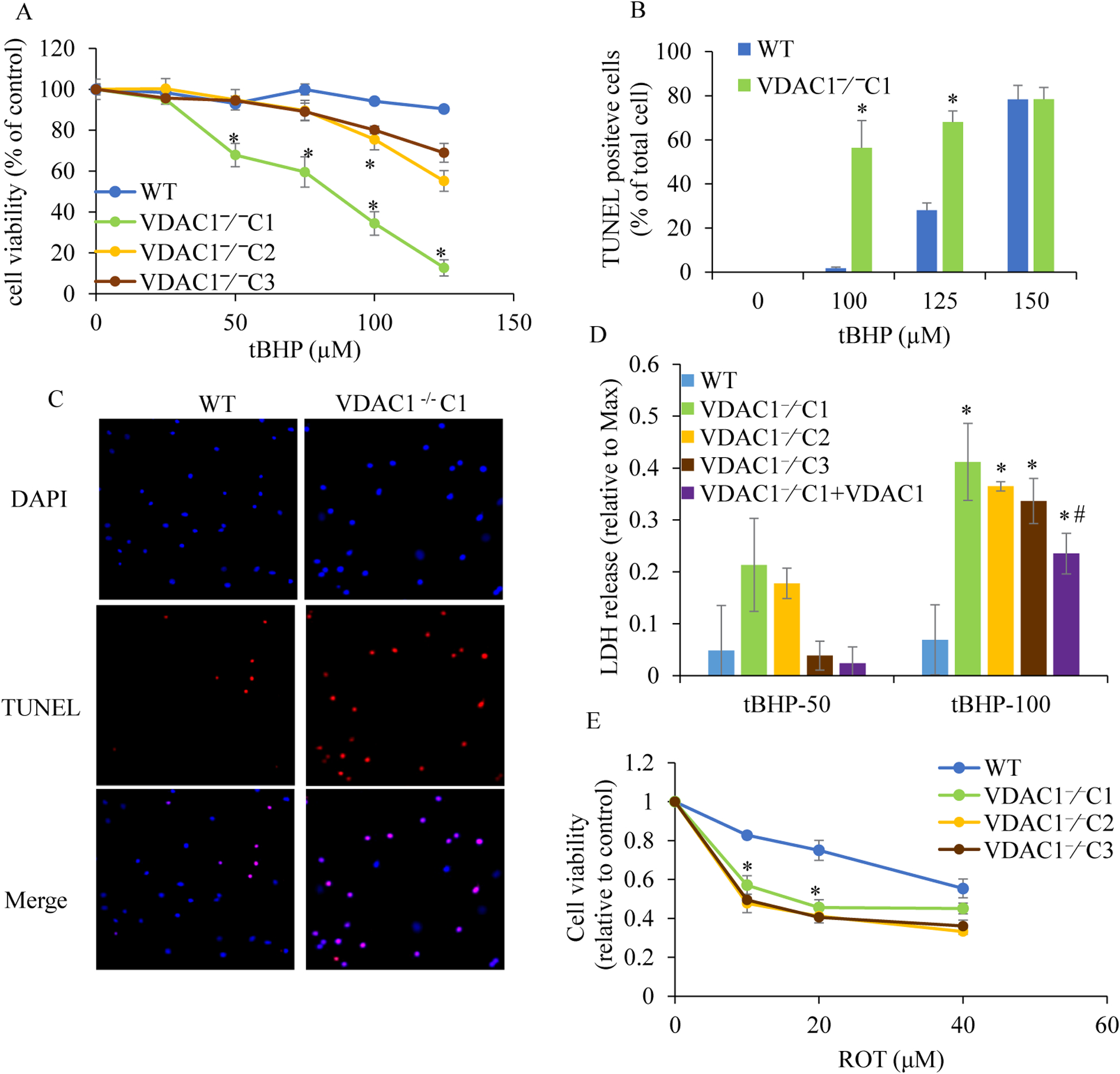
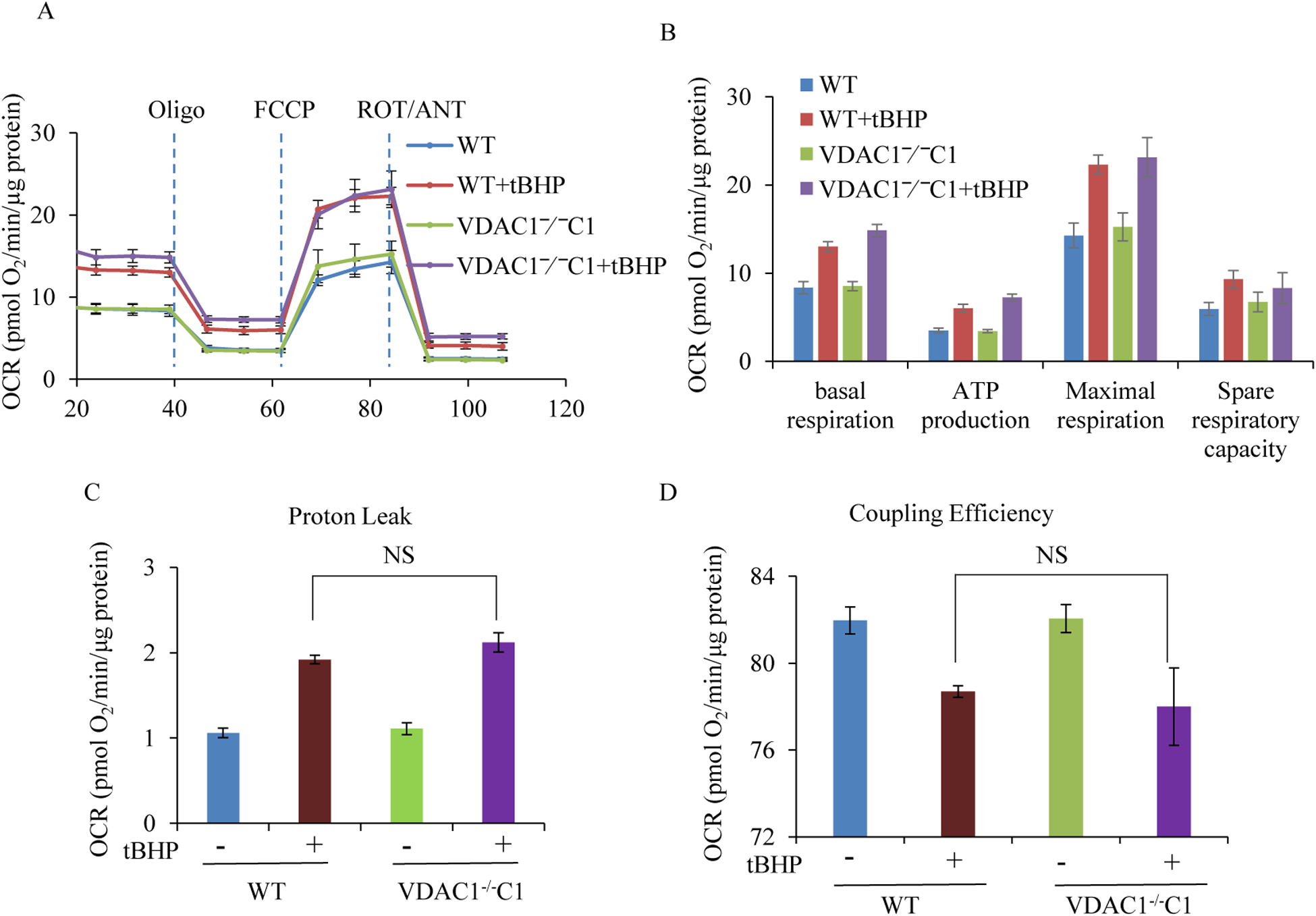
We knocked out VDAC1 in this rat cardiomyoblast cell line with CRISPR-Cas9 genome editing technique to produce VDAC1 H9c2 cells, and determined if VDAC1 is critical in promoting cell death via oxidative stress induced by tert-butylhydroperoxide (tBHP), an organic peroxide, or rotenone (ROT), an inhibitor of mitochondrial complex I by measuring cell viability with MTT assay, cell death with TUNEL stain and LDH release. The mitochondrial and glycolytic stress were examined by measuring O consumption rate (OCR) and extracellular acidification rate (ECAR) with a Seahorse XFp analyzer.
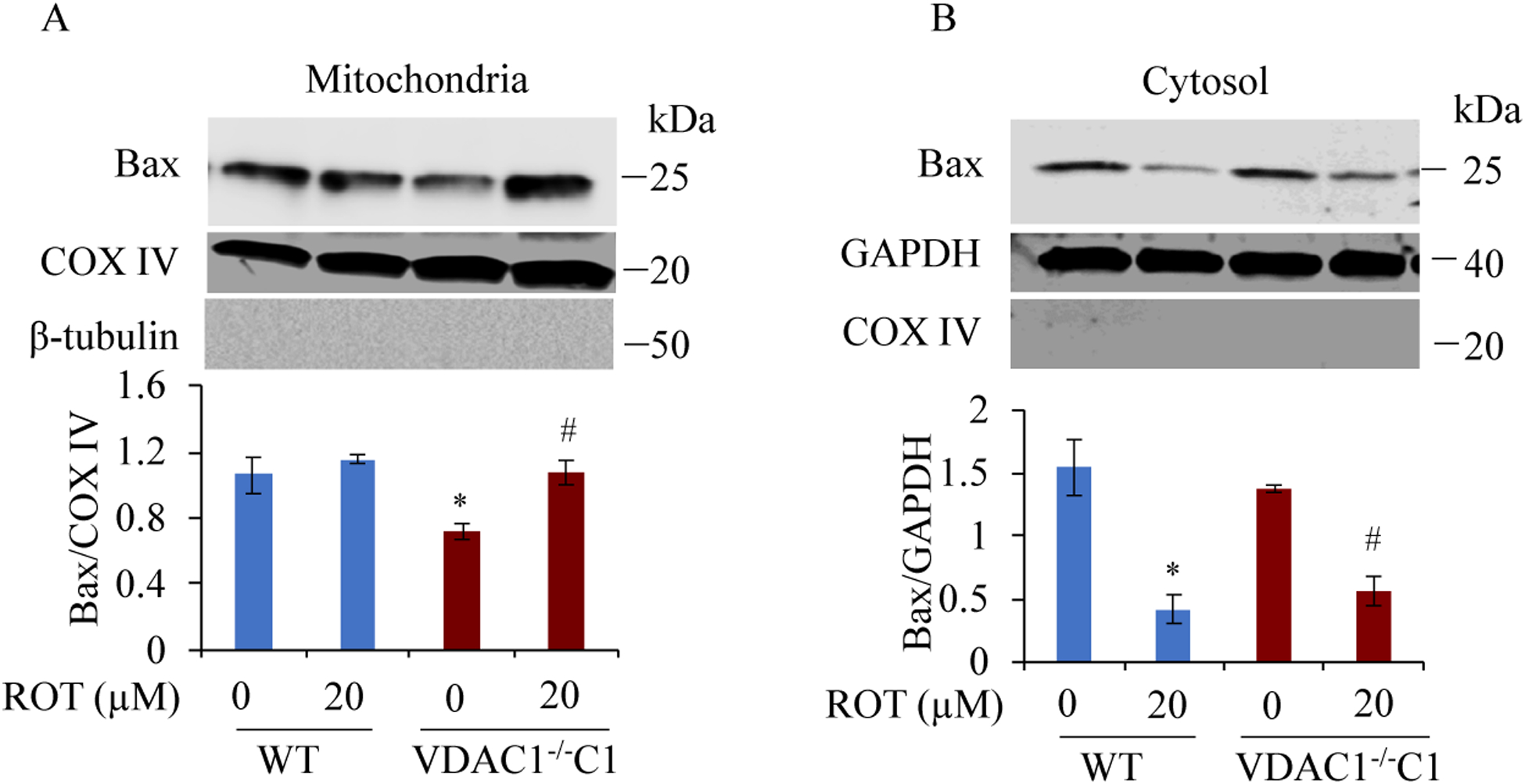
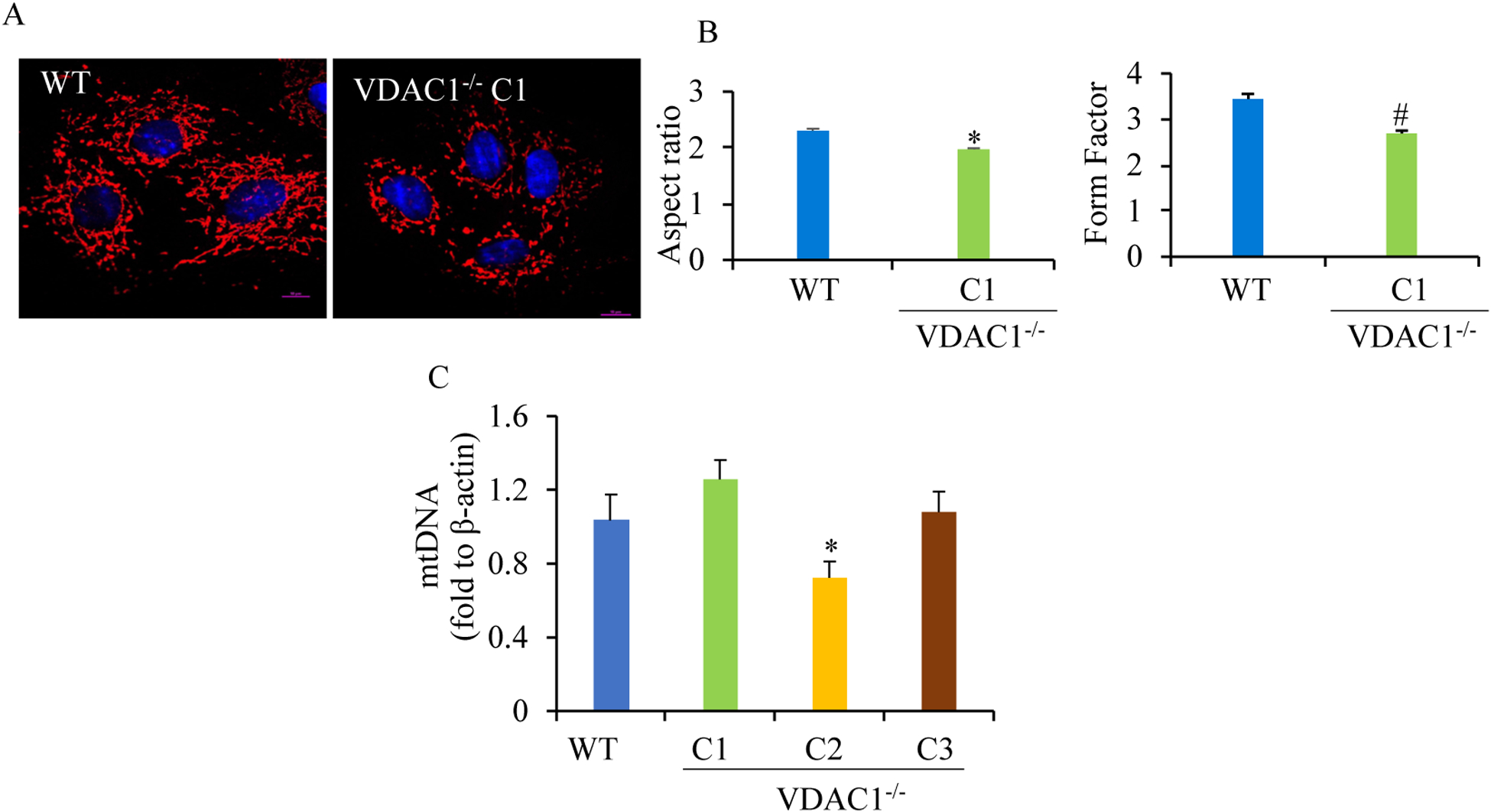
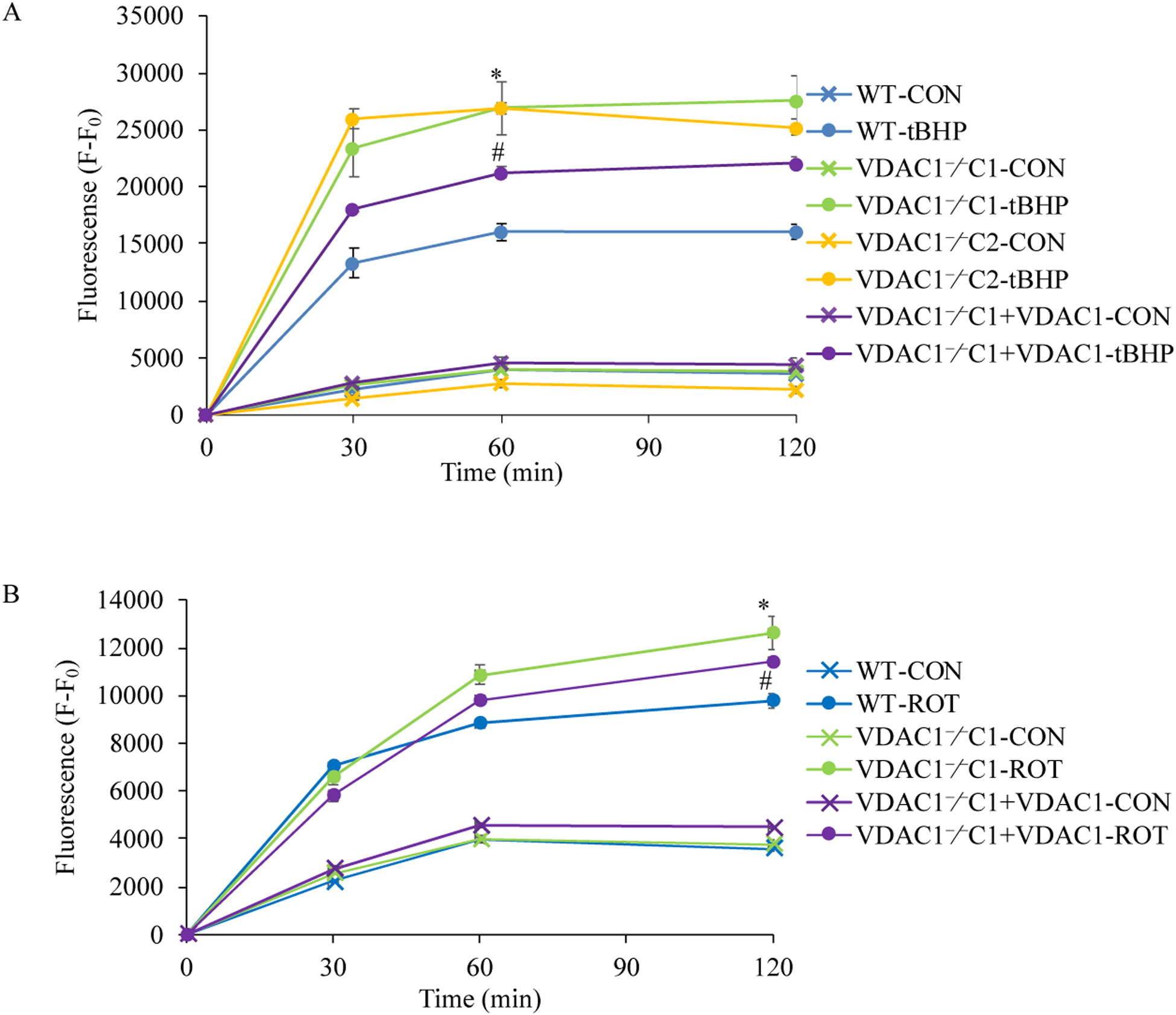
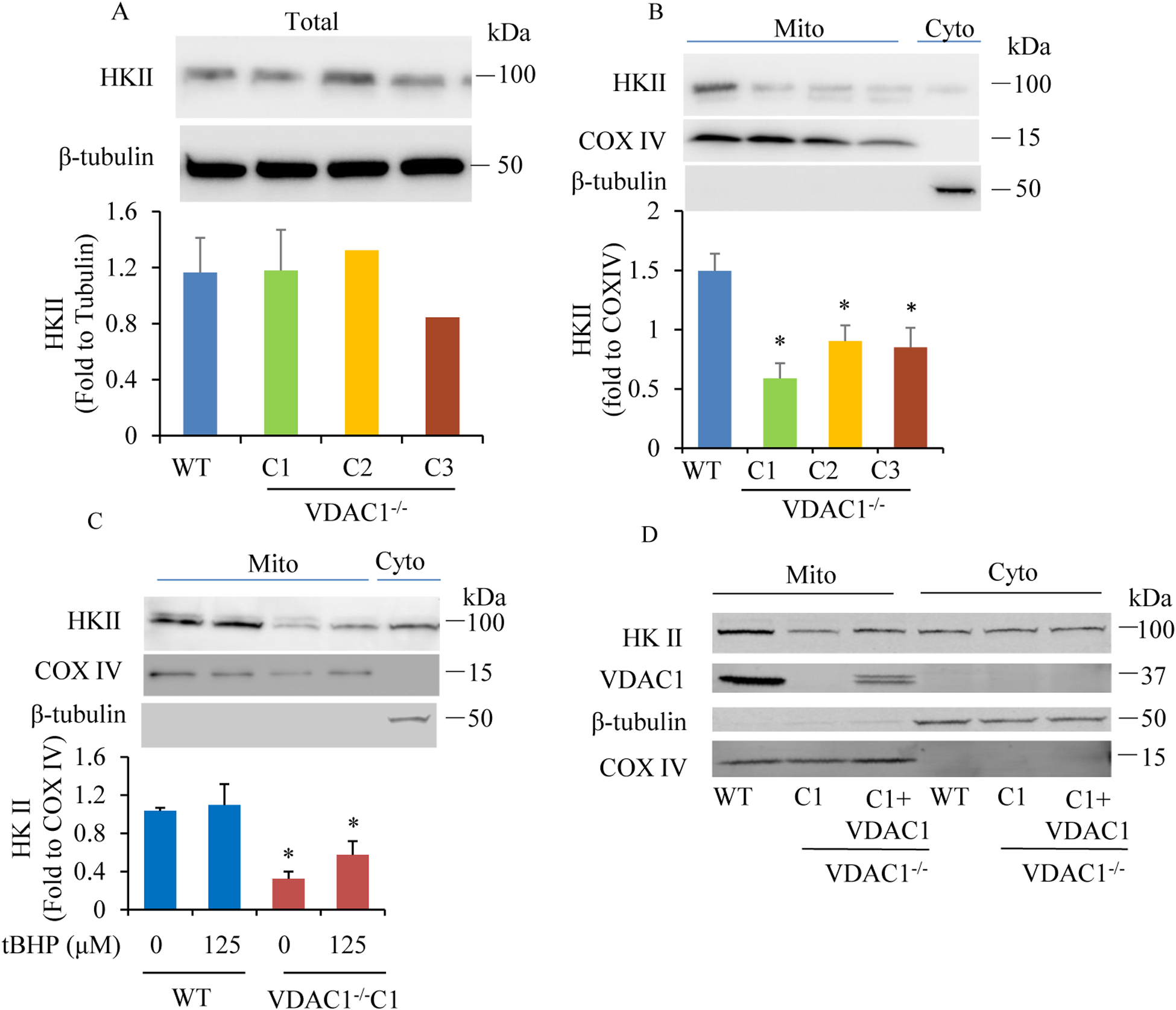
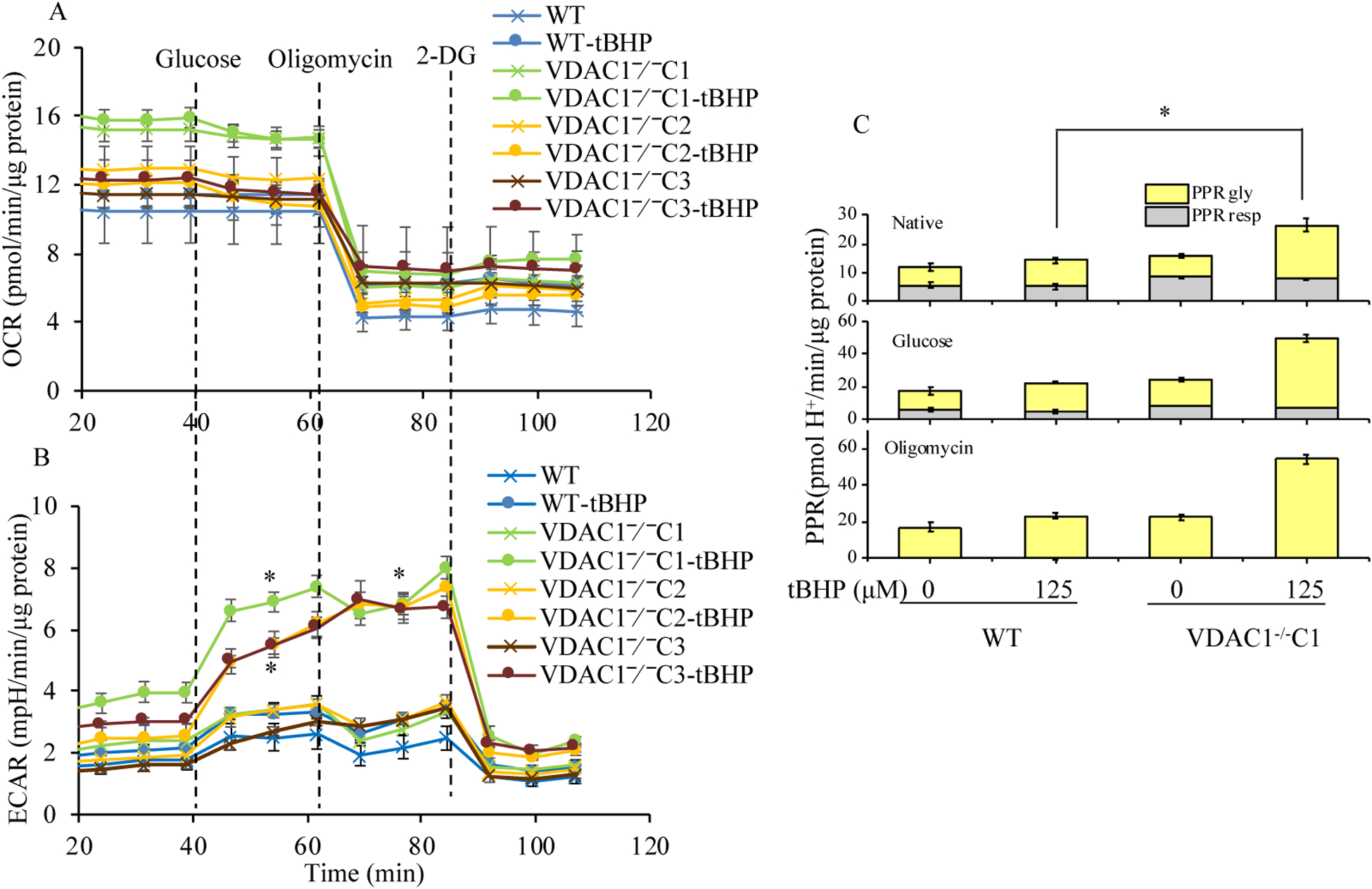
We found that under control conditions, VDAC1 did not affect H9c2 cell proliferation or mitochondrial respiration. However, compared to the wildtype (WT) cells, exposure to either tBHP or ROT enhanced the production of ROS, ECAR, and the proton (H) production rate (PPR) from glycolysis, as well as promoted apoptotic cell death in VDAC1 H9c2 cells. VDAC1 H9c2 cells also exhibited markedly reduced mitochondria-bound hexokinase II (HKII) and Bax. Restoration of VDAC1 in VDAC1 H9c2 cells reinstated mitochondria-bound HKII and concomitantly decreased tBHP and ROT-induced ROS production and cell death. Interestingly, mitochondrial respiration remained the same after tBHP treatment in VDAC1 and WT H9c2 cells.
Our results suggest that VDAC1 in H9c2 cells enhances oxidative stress-mediated cell apoptosis that is directly linked to the reduction of mitochondria-bound HKII and concomitantly associated with enhanced ROS production, ECAR, and PPR.
背景/目的:电压依赖性阴离子通道 1(VDAC1)是最丰富的线粒体外膜蛋白,其在细胞死亡中的作用取决于细胞类型和刺激物。在各种类型的癌细胞系中,VDAC1 的沉默和上调均可刺激细胞凋亡。相比之下,在小鼠胚胎干细胞(MES)和小鼠胚胎成纤维细胞(MEF)中,VDAC1 敲除(VDAC1)在凋亡性细胞死亡中的作用是矛盾的。VDAC1 在心脏细胞氧化应激诱导的细胞死亡中的贡献和潜在机制尚未建立。我们假设 VDAC1 是 H9c2 细胞氧化应激诱导细胞死亡的重要调节因子。
我们使用 CRISPR-Cas9 基因组编辑技术敲除该大鼠心肌细胞系中的 VDAC1,以产生 VDAC1 H9c2 细胞,并通过 MTT 测定法测量细胞活力、TUNEL 染色和 LDH 释放来确定 VDAC1 是否通过 tert-butylhydroperoxide(tBHP)诱导的氧化应激或 Rotenone(ROT)促进细胞死亡至关重要,后者是一种线粒体复合物 I 抑制剂。使用 Seahorse XFp 分析仪测量耗氧量(OCR)和细胞外酸化率(ECAR)来检测线粒体和糖酵解应激。
我们发现,在对照条件下,VDAC1 不会影响 H9c2 细胞的增殖或线粒体呼吸。然而,与野生型(WT)细胞相比,暴露于 tBHP 或 ROT 会增强 ROS、ECAR 和糖酵解产生的质子(H)产生率(PPR),并促进 VDAC1 H9c2 细胞的凋亡性细胞死亡。VDAC1 H9c2 细胞还表现出明显减少的线粒体结合己糖激酶 II(HKII)和 Bax。在 VDAC1 H9c2 细胞中恢复 VDAC1 可恢复线粒体结合的 HKII,同时降低 tBHP 和 ROT 诱导的 ROS 产生和细胞死亡。有趣的是,在 VDAC1 和 WT H9c2 细胞中 tBHP 处理后线粒体呼吸保持不变。
我们的结果表明,H9c2 细胞中的 VDAC1 增强了氧化应激介导的细胞凋亡,这直接与线粒体结合的 HKII 的减少有关,并与 ROS 产生、ECAR 和 PPR 的增加相关。