Department of Mathematics, University of Michigan, 530 Church Street, Ann Arbor, 48109, USA.
Department of Mathematics, Yale University, 10 Hillhouse Ave, New Haven, 06511, USA.
BMC Bioinformatics. 2020 Oct 23;21(1):477. doi: 10.1186/s12859-020-03641-z.
High throughput microfluidic protocols in single cell RNA sequencing (scRNA-seq) collect mRNA counts from up to one million individual cells in a single experiment; this enables high resolution studies of rare cell types and cell development pathways. Determining small sets of genetic markers that can identify specific cell populations is thus one of the major objectives of computational analysis of mRNA counts data. Many tools have been developed for marker selection on single cell data; most of them, however, are based on complex statistical models and handle the multi-class case in an ad-hoc manner.
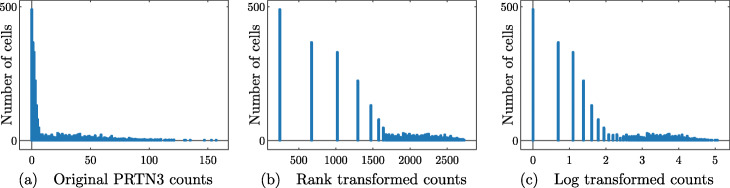
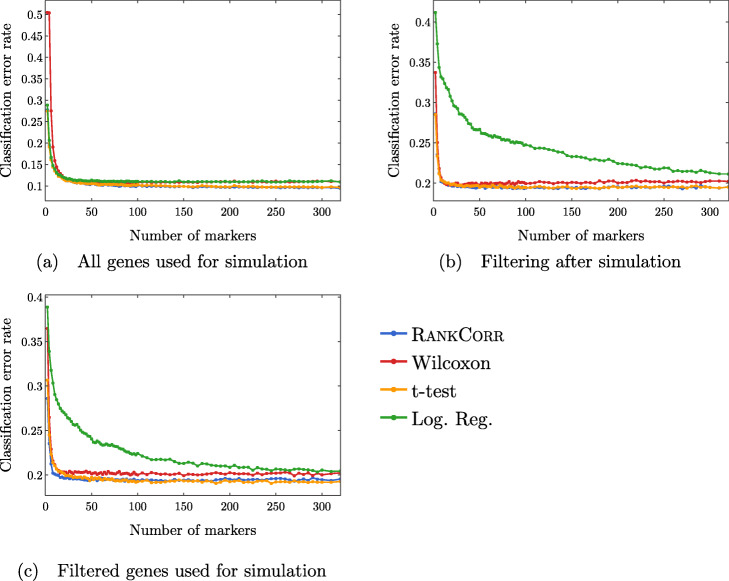
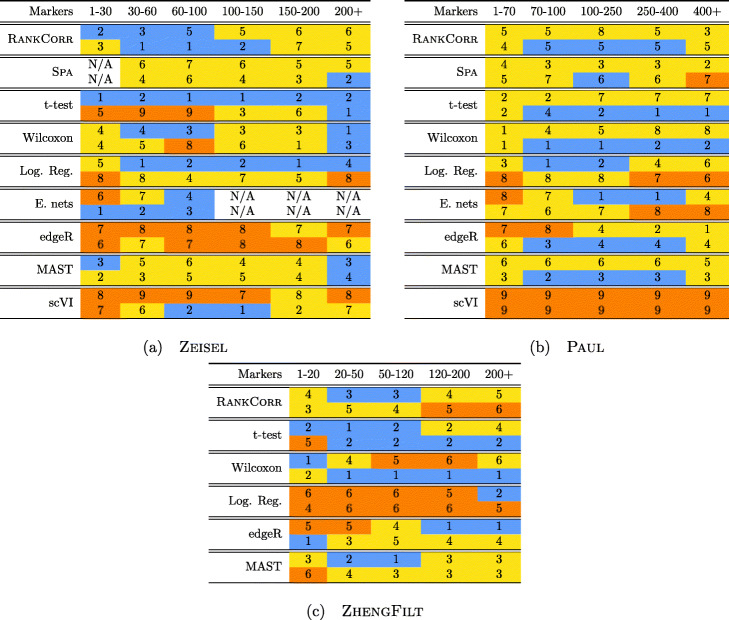
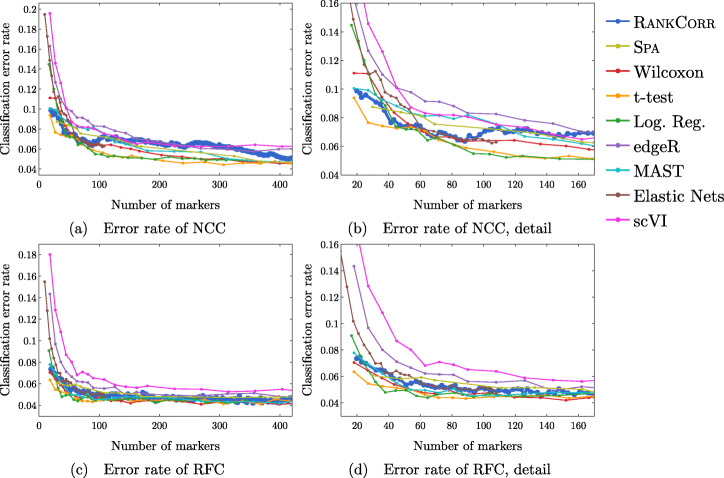
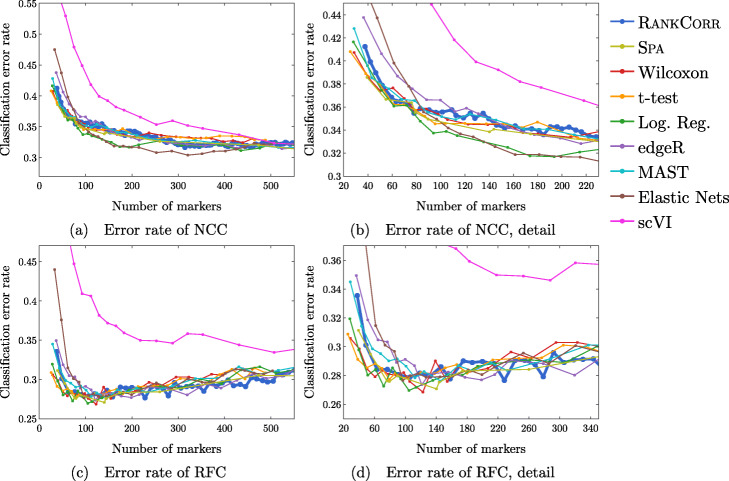
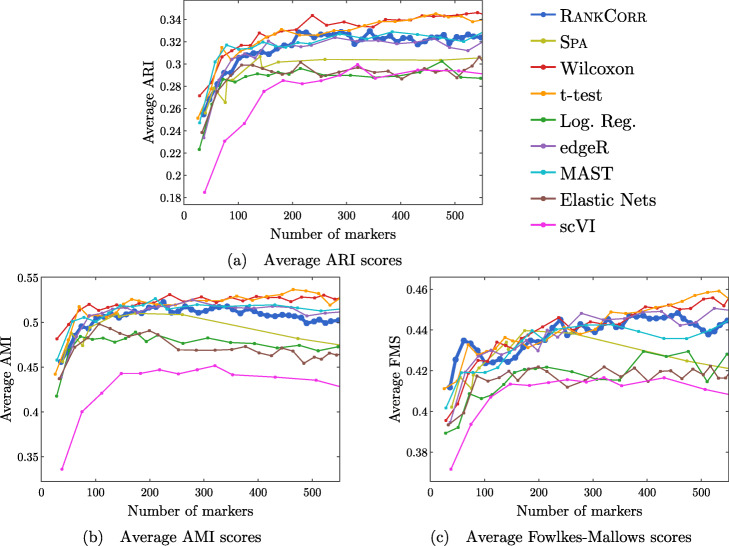
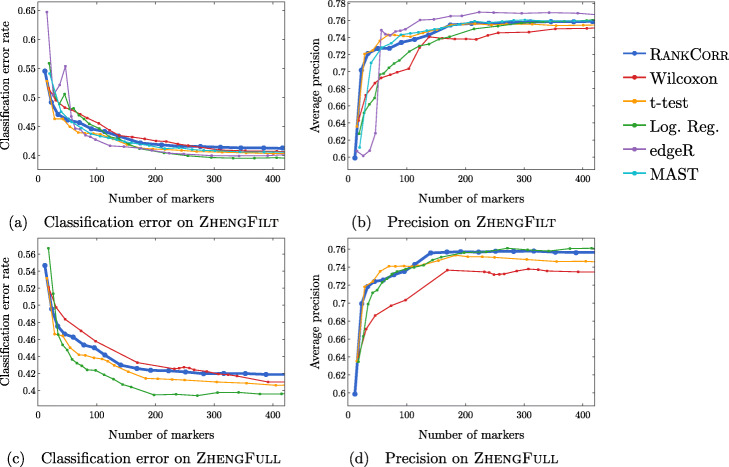
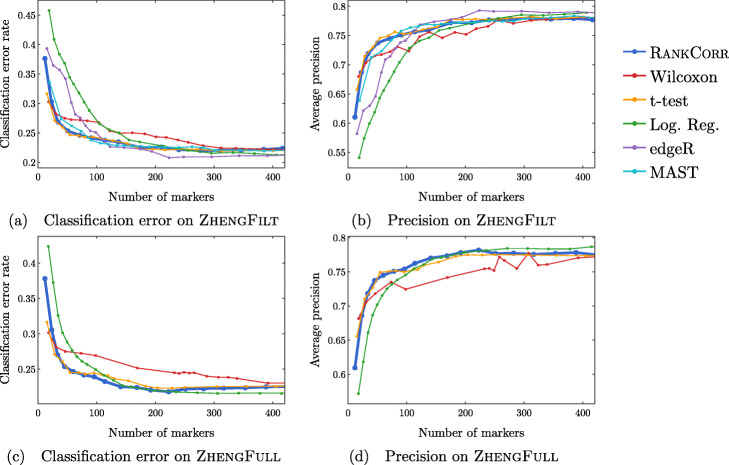
We introduce RANKCORR, a fast method with strong mathematical underpinnings that performs multi-class marker selection in an informed manner. RANKCORR proceeds by ranking the mRNA counts data before linearly separating the ranked data using a small number of genes. The step of ranking is intuitively natural for scRNA-seq data and provides a non-parametric method for analyzing count data. In addition, we present several performance measures for evaluating the quality of a set of markers when there is no known ground truth. Using these metrics, we compare the performance of RANKCORR to a variety of other marker selection methods on an assortment of experimental and synthetic data sets that range in size from several thousand to one million cells.
According to the metrics introduced in this work, RANKCORR is consistently one of most optimal marker selection methods on scRNA-seq data. Most methods show similar overall performance, however; thus, the speed of the algorithm is the most important consideration for large data sets (and comparing the markers selected by several methods can be fruitful). RANKCORR is fast enough to easily handle the largest data sets and, as such, it is a useful tool to add into computational pipelines when dealing with high throughput scRNA-seq data. RANKCORR software is available for download at https://github.com/ahsv/RankCorr with extensive documentation.
高通量微流控方案在单细胞 RNA 测序(scRNA-seq)中,在单个实验中收集多达一百万个别细胞的 mRNA 计数;这使得对稀有细胞类型和细胞发育途径进行高分辨率研究成为可能。因此,确定可以识别特定细胞群体的小遗传标记集是 scRNA-seq 数据的计算分析的主要目标之一。已经开发了许多用于单细胞数据标记选择的工具;然而,其中大多数都是基于复杂的统计模型,并以特定的方式处理多类情况。
我们引入了 RANKCORR,这是一种快速的方法,具有强大的数学基础,可以以明智的方式进行多类标记选择。RANKCORR 通过对 mRNA 计数数据进行排序,然后使用少量基因对排序后的数据进行线性分离来进行操作。排序步骤对于 scRNA-seq 数据来说是直观自然的,并且为分析计数数据提供了一种非参数方法。此外,我们提出了几种性能指标,用于在没有已知真实情况的情况下评估一组标记的质量。使用这些指标,我们在各种大小从几千到一百万细胞的实验和合成数据集上比较了 RANKCORR 与其他各种标记选择方法的性能。
根据本工作中引入的指标,RANKCORR 在 scRNA-seq 数据上始终是最优化的标记选择方法之一。然而,大多数方法的整体性能相似;因此,对于大型数据集来说,算法的速度是最重要的考虑因素(比较几种方法选择的标记可能会很有成果)。RANKCORR 足够快,可以轻松处理最大的数据集,因此在处理高通量 scRNA-seq 数据时,它是计算管道中有用的工具。RANKCORR 软件可在 https://github.com/ahsv/RankCorr 下载,附带详细文档。