Department of Computer Science and Technology, University of Cambridge, Cambridge, UK.
Department of Applied Mathematics and Statistics, Johns Hopkins University, Baltimore, MD, USA.
Nat Commun. 2021 Feb 19;12(1):1186. doi: 10.1038/s41467-021-21453-4.
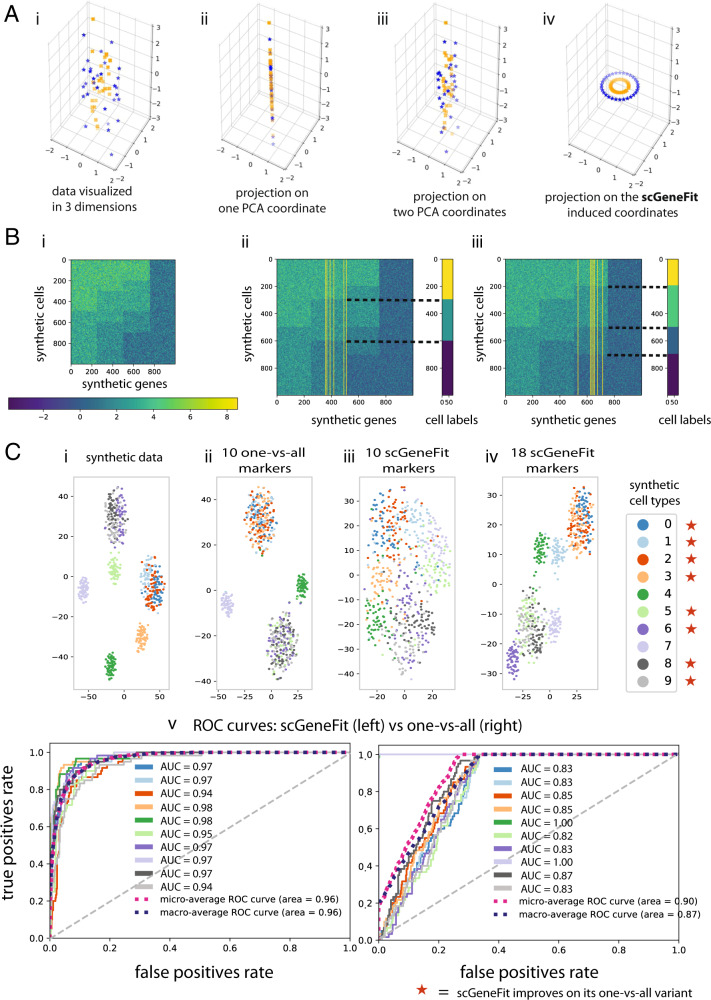
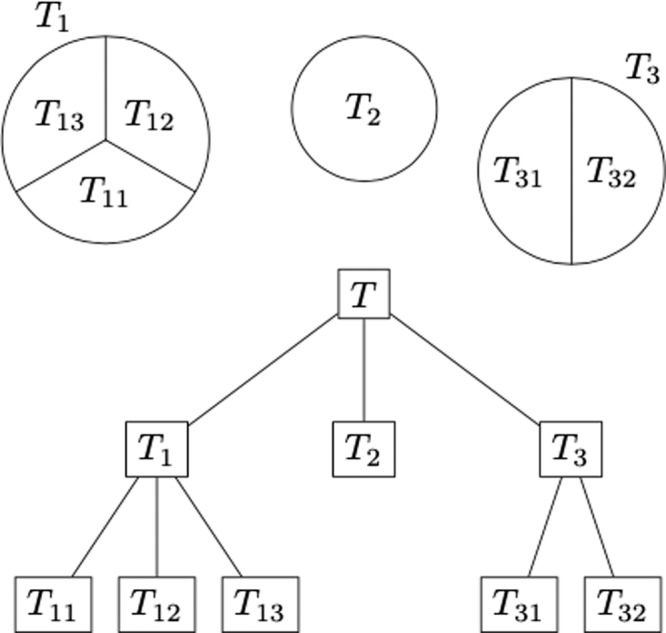
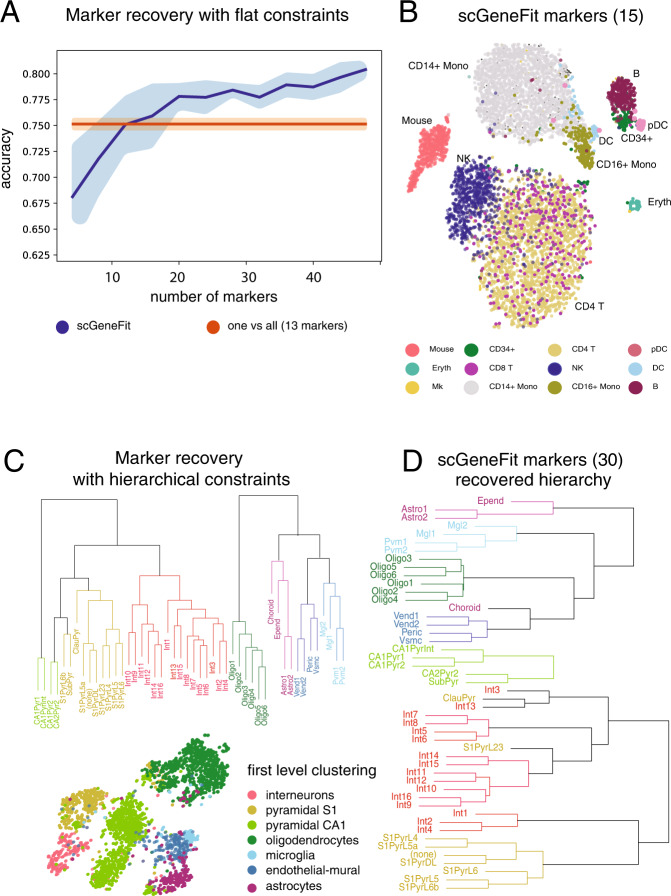
Single-cell technologies characterize complex cell populations across multiple data modalities at unprecedented scale and resolution. Multi-omic data for single cell gene expression, in situ hybridization, or single cell chromatin states are increasingly available across diverse tissue types. When isolating specific cell types from a sample of disassociated cells or performing in situ sequencing in collections of heterogeneous cells, one challenging task is to select a small set of informative markers that robustly enable the identification and discrimination of specific cell types or cell states as precisely as possible. Given single cell RNA-seq data and a set of cellular labels to discriminate, scGeneFit selects gene markers that jointly optimize cell label recovery using label-aware compressive classification methods. This results in a substantially more robust and less redundant set of markers than existing methods, most of which identify markers that separate each cell label from the rest. When applied to a data set given a hierarchy of cell types as labels, the markers found by our method improves the recovery of the cell type hierarchy with fewer markers than existing methods using a computationally efficient and principled optimization.
单细胞技术以前所未有的规模和分辨率描绘了多个数据模态的复杂细胞群体。单细胞基因表达、原位杂交或单细胞染色质状态的多组学数据在不同的组织类型中越来越多地得到应用。当从分离的细胞样本中分离特定的细胞类型或在异质细胞的集合中进行原位测序时,一个具有挑战性的任务是选择一小部分信息丰富的标记物,这些标记物能够稳健地尽可能精确地识别和区分特定的细胞类型或细胞状态。给定单细胞 RNA-seq 数据和一组要区分的细胞标签,scGeneFit 使用标签感知压缩分类方法选择共同优化细胞标签恢复的基因标记物。这导致了比现有方法更稳健、更少冗余的标记物集,而大多数现有方法识别的标记物将每个细胞标签与其他标签分开。当应用于给定细胞类型层次结构作为标签的数据集时,与使用现有方法相比,我们的方法发现的标记物使用计算效率高且有原则的优化以更少的标记物提高了细胞类型层次结构的恢复。