Chair of Theoretical Chemistry and Catalysis Research Center, Technische Universität München, Garching, Germany.
Engineering Laboratory, University of Cambridge, Cambridge, CB2 1PZ, UK.
Nat Commun. 2020 Oct 30;11(1):5505. doi: 10.1038/s41467-020-19267-x.
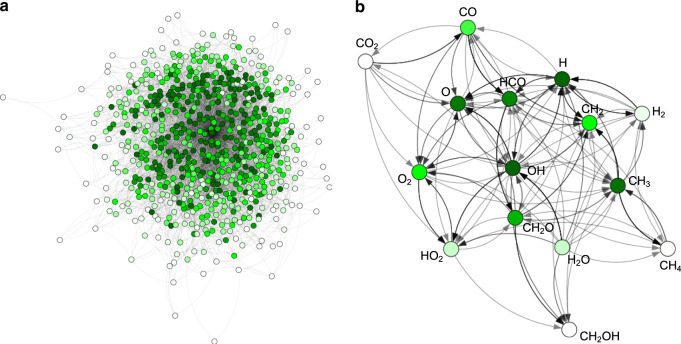
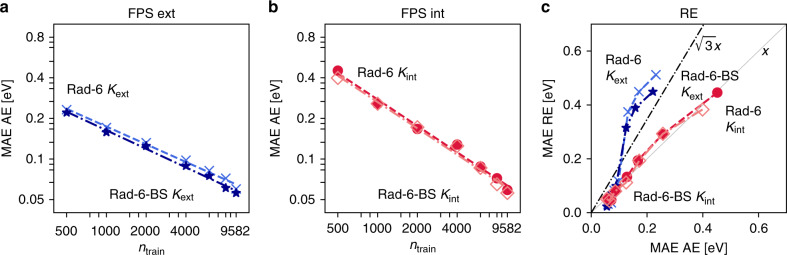
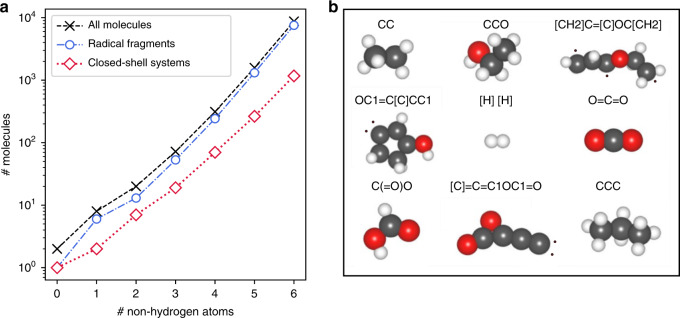
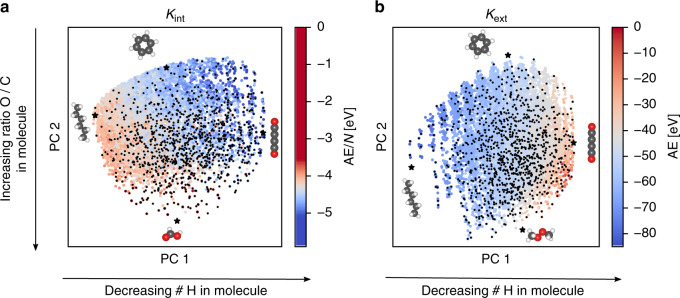
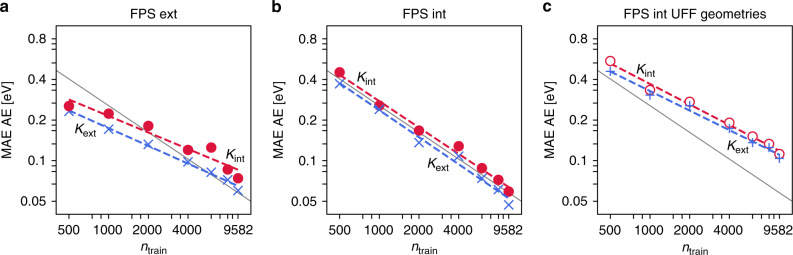
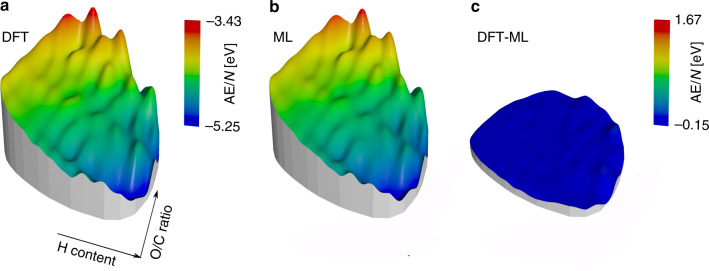
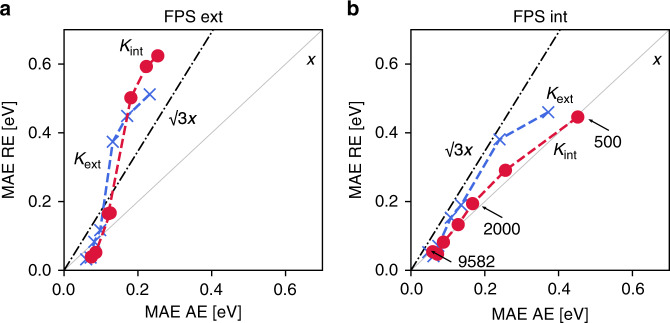
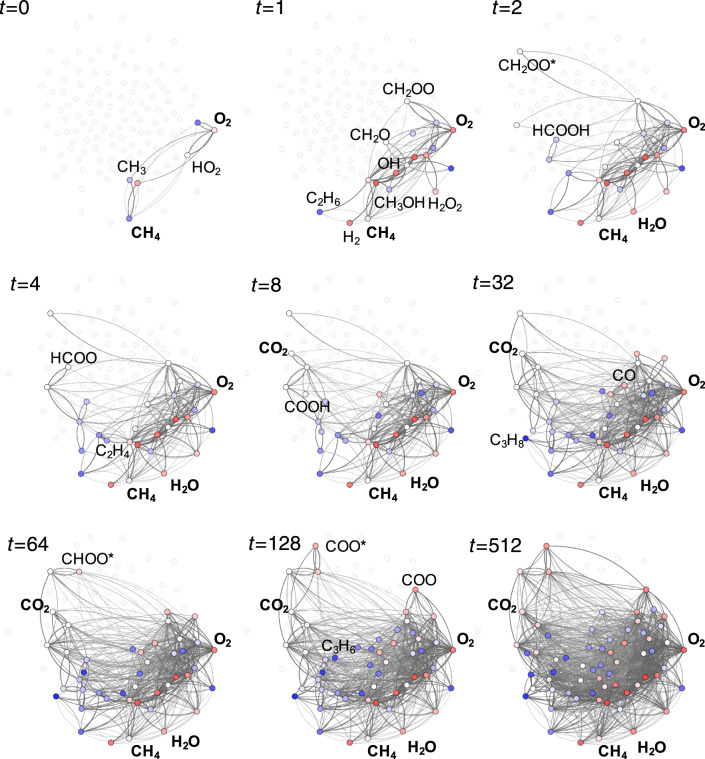
Chemical compound space refers to the vast set of all possible chemical compounds, estimated to contain 10 molecules. While intractable as a whole, modern machine learning (ML) is increasingly capable of accurately predicting molecular properties in important subsets. Here, we therefore engage in the ML-driven study of even larger reaction space. Central to chemistry as a science of transformations, this space contains all possible chemical reactions. As an important basis for 'reactive' ML, we establish a first-principles database (Rad-6) containing closed and open-shell organic molecules, along with an associated database of chemical reaction energies (Rad-6-RE). We show that the special topology of reaction spaces, with central hub molecules involved in multiple reactions, requires a modification of existing compound space ML-concepts. Showcased by the application to methane combustion, we demonstrate that the learned reaction energies offer a non-empirical route to rationally extract reduced reaction networks for detailed microkinetic analyses.
化学化合物空间是指所有可能的化学化合物的广阔集合,据估计包含 10^90 个分子。虽然作为一个整体是难以处理的,但现代机器学习(ML)越来越能够准确地预测重要子集的分子性质。在这里,我们因此从事更大的反应空间的 ML 驱动研究。作为化学作为转化科学的核心,这个空间包含了所有可能的化学反应。作为“反应性”ML 的重要基础,我们建立了一个包含闭壳和开壳有机分子的第一性原理数据库(Rad-6),以及一个相关的化学反应能数据库(Rad-6-RE)。我们表明,反应空间的特殊拓扑结构,其中中心枢纽分子参与多个反应,需要对现有的化合物空间 ML 概念进行修改。通过对甲烷燃烧的应用,我们证明了所学习的反应能为合理提取用于详细微观动力学分析的简化反应网络提供了一条非经验途径。