Chen Yi-Hau, Wang Hsiuying
Institute of Statistical Science, Academia Sinica, Nankang, Taipei, Taiwan.
Institute of Statistics, National Chiao Tung University, Hsinchu, Taiwan.
Infect Drug Resist. 2020 Oct 29;13:3887-3894. doi: 10.2147/IDR.S277620. eCollection 2020.
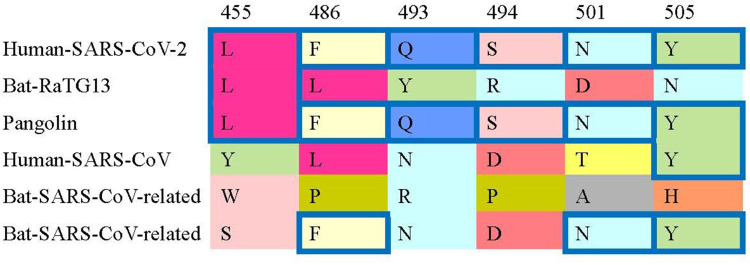
The number of COVID-19 infections worldwide has reached 10 million. COVID‑19 caused by SARS-CoV-2 is more contagious than SARS-CoV-1. There is a dispute about the origin of COVID-19. Study results showed that all SARS-CoV-2 sequences around the world share a common ancestor towards the end of 2019.
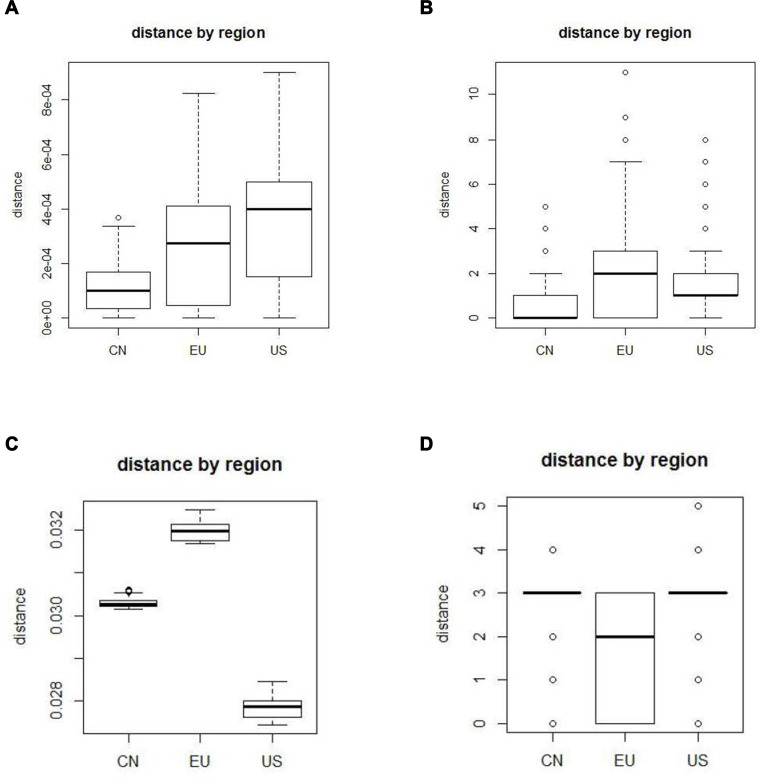
Virus sequences from COVID-19 samples at the early time should be less diversifiable than those from samples at the later time because there might be more mutations when the virus evolutes over time. The diversity of virus nucleotide sequences can be measured by the nucleotide substitution distance. To explore the diversity of SARS-CoV-2, we use different nucleotide substitution models to calculate the distances of SARS-CoV-2 samples from 3 different areas, China, Europe, and the USA. Then, we use these distances to infer the origin of COVID-19.
It is known that COVID-19 originated in Wuhan China and then spread to Europe and the USA. By using different substitution models, the distances of SARS-CoV-2 samples from these areas are significantly different. By ANOVA testing, the p-value is less than 2.2e-16. The analyzed results in most substitution models show that China has the lowest diversity, followed by Europe and lastly by the USA. This outcome coincides with the virus transmission time order that SARS-CoV-2 starts in China, then outbreaks in Europe and finally in the USA.
The magnitude of nucleotide substitution distance of SARS-CoV-2 is closely related to the transmission time order of SARS-CoV-2. This outcome reveals that the nucleotide substitution distance of SARS-CoV-2 may be used to infer the origin of COVID-19.
全球新冠病毒感染病例数已达1000万。严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引起的新冠病毒比严重急性呼吸综合征冠状病毒1(SARS-CoV-1)传染性更强。关于新冠病毒的起源存在争议。研究结果表明,全球所有SARS-CoV-2序列在2019年底有一个共同祖先。
由于病毒随时间进化可能会出现更多突变,新冠病毒早期样本的病毒序列多样性应低于后期样本。病毒核苷酸序列的多样性可以通过核苷酸替代距离来衡量。为了探究SARS-CoV-2的多样性,我们使用不同的核苷酸替代模型来计算来自中国、欧洲和美国3个不同地区的SARS-CoV-2样本的距离。然后,我们利用这些距离来推断新冠病毒的起源。
已知新冠病毒起源于中国武汉,然后传播到欧洲和美国。通过使用不同的替代模型,来自这些地区的SARS-CoV-2样本的距离存在显著差异。通过方差分析测试,p值小于2.2×10⁻¹⁶。大多数替代模型的分析结果表明,中国的多样性最低,其次是欧洲,最后是美国。这一结果与SARS-CoV-2先在中国出现,然后在欧洲爆发,最后在美国爆发的病毒传播时间顺序一致。
SARS-CoV-2的核苷酸替代距离大小与SARS-CoV-2的传播时间顺序密切相关。这一结果表明,SARS-CoV-2的核苷酸替代距离可用于推断新冠病毒的起源。