Bautista Mary Ann C, Zheng Yan, Hu Zhangli, Deng Yunfei, Chen Tao
South China Botanical Garden, Chinese Academy of Sciences, Guangzhou 510650, China.
Fairy Lake Botanical Garden, Chinese Academy of Sciences, Shenzhen 518004, China.
Plants (Basel). 2020 Nov 28;9(12):1671. doi: 10.3390/plants9121671.
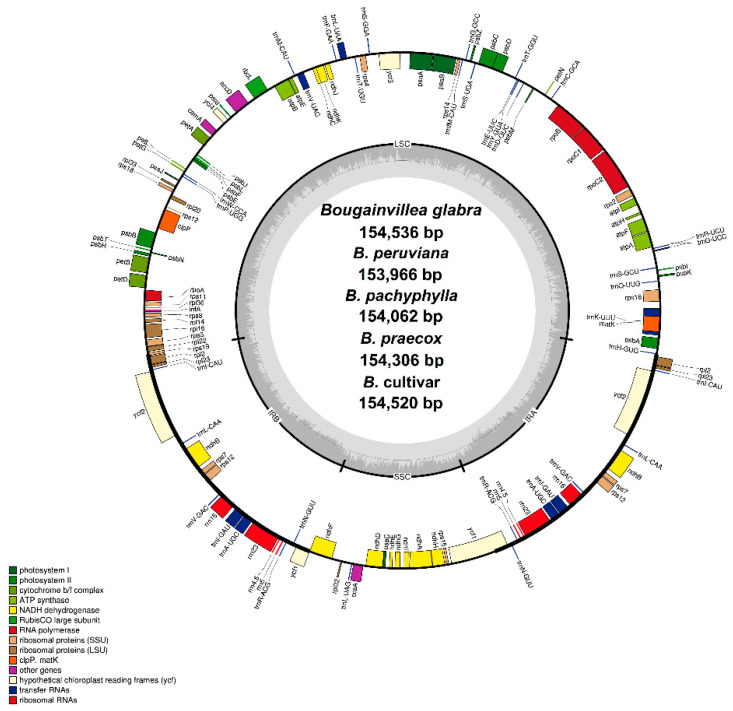
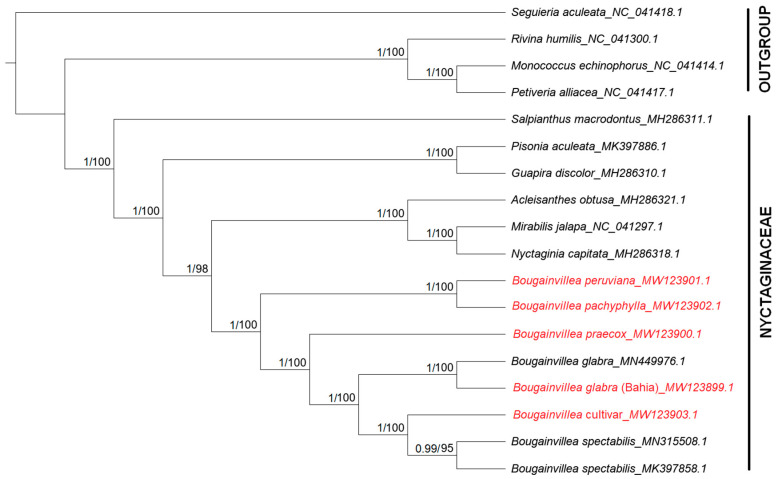
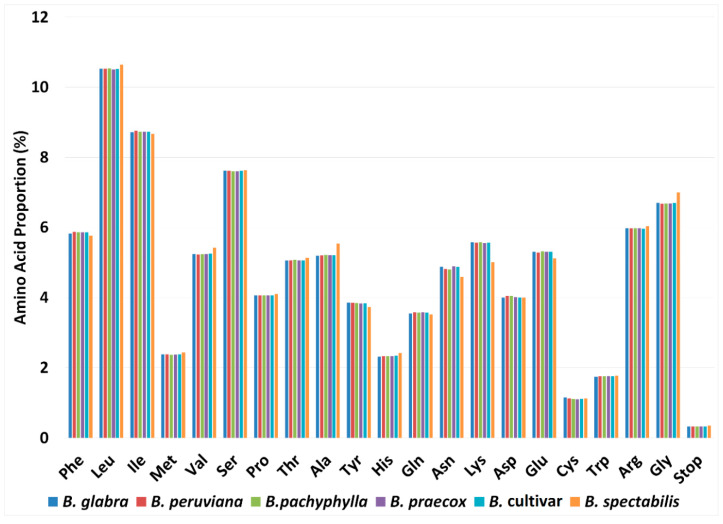
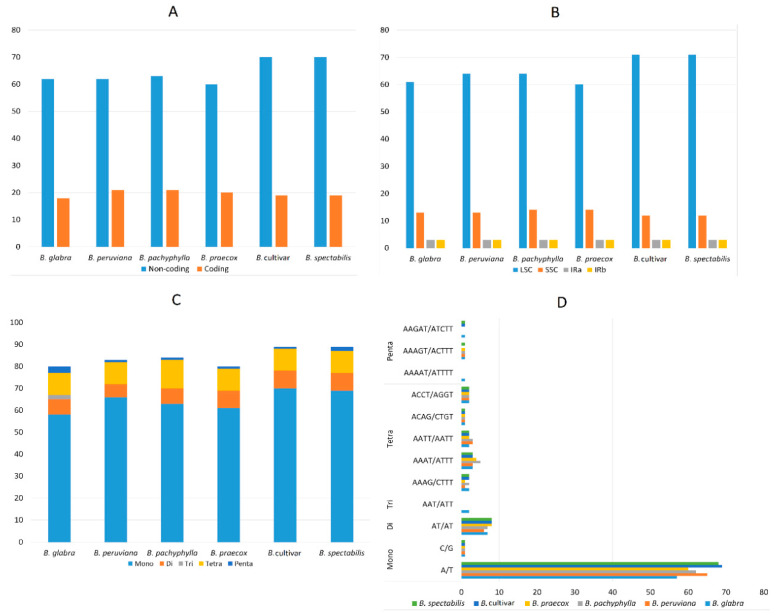
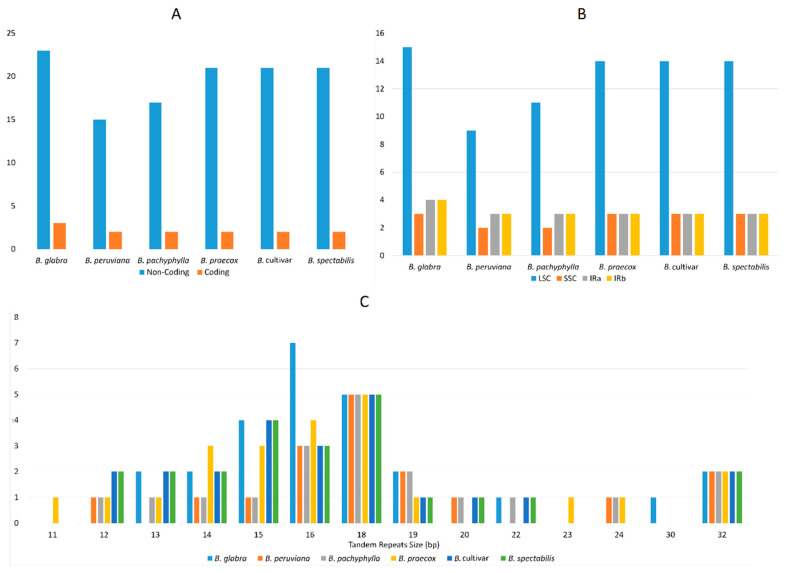
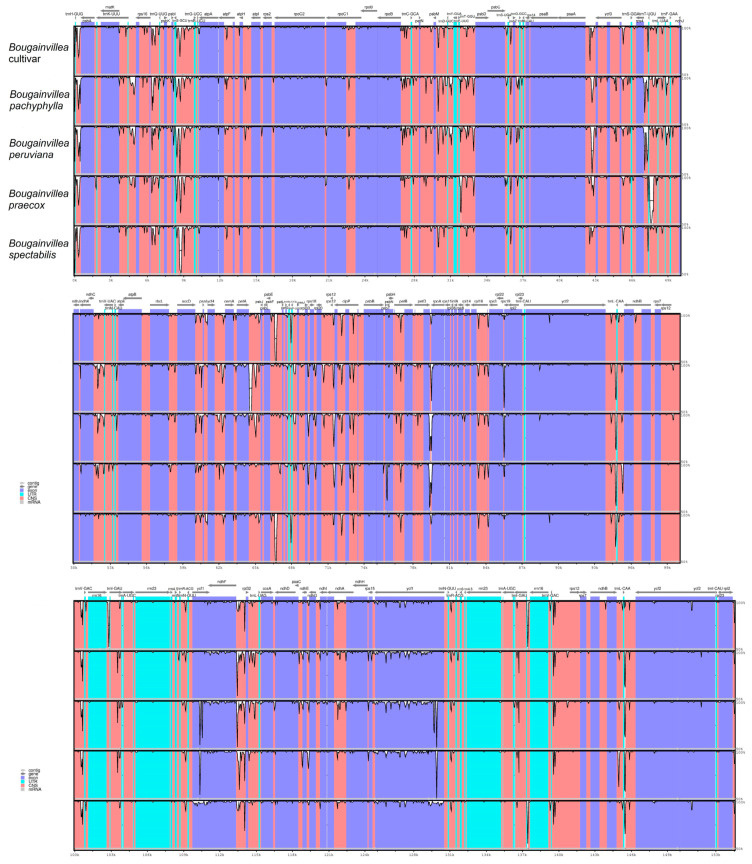
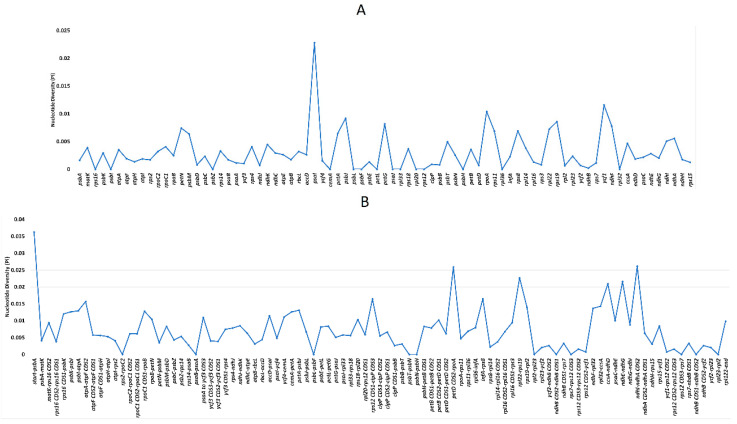
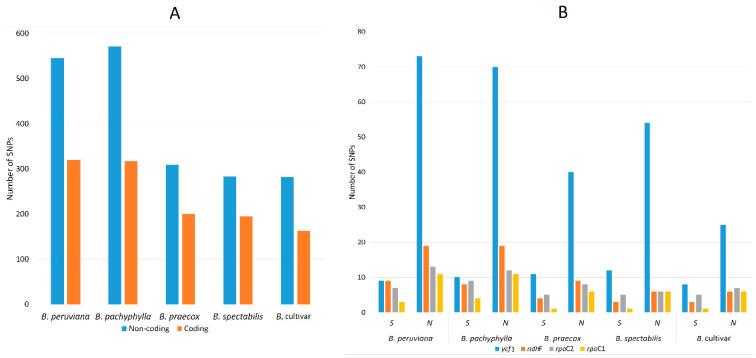
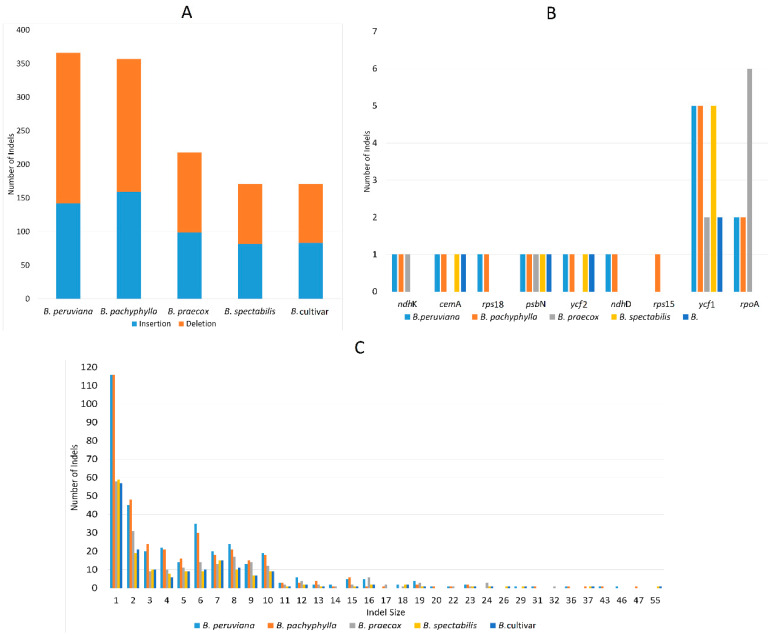
(Nyctaginaceae) is a popular ornamental plant group primarily grown for its striking colorful bracts. However, despite its established horticultural value, limited genomic resources and molecular studies have been reported for this genus. Thus, to address this existing gap, complete chloroplast genomes of four species ( ) and one cultivar were sequenced and characterized. The cp genomes range from 153,966 bp to 154,541 bp in length, comprising a large single-copy region (85,159 bp-85,708 bp) and a small single-copy region (18,014 bp-18,078 bp) separated by a pair of inverted repeats (25,377-25,427 bp). All sequenced plastomes have 131 annotated genes, including 86 protein-coding, eight rRNA, and 37 tRNA genes. These five newly sequenced cp genomes were compared to the cp genome deposited in GeBank. The results showed that all cp genomes have highly similar structures, contents, and organization. They all exhibit quadripartite structures and all have the same numbers of genes and introns. Codon usage, RNA editing sites, and repeat analyses also revealed highly similar results for the six cp genomes. The amino acid leucine has the highest proportion and almost all favored synonymous codons have either an A or U ending. Likewise, out of the 42 predicted RNA sites, most conversions were from serine (S) to leucine (L). The majority of the simple sequence repeats detected were A/T mononucleotides, making the cp genomes A/T-rich. The contractions and expansions of the IR boundaries were very minimal as well, hence contributing very little to the differences in genome size. In addition, sequence variation analyses showed that cp genomes share nearly identical genomic profiles though several potential barcodes, such as 1, F, and A were identified. Higher variation was observed in both and cp sequences based on SNPs and indels analysis. Phylogenetic reconstructions further showed that these two species appear to be the basal taxa of . The rarely cultivated and wild species of ( ) diverged earlier than the commonly cultivated species and cultivar ( cv.). Overall, the results of this study provide additional genetic resources that can aid in further phylogenetic and evolutionary studies in . Moreover, genetic information from this study is potentially useful in identifying species and cultivars, which is essential for both taxonomic and plant breeding studies.
紫茉莉科是一类广受欢迎的观赏植物,主要因其引人注目的彩色苞片而种植。然而,尽管其具有既定的园艺价值,但关于该属的基因组资源和分子研究报道有限。因此,为了填补这一现有空白,对四个物种( )和一个品种的完整叶绿体基因组进行了测序和特征分析。这些叶绿体基因组长度在153,966 bp至154,541 bp之间,包括一个大单拷贝区域(85,159 bp - 85,708 bp)和一个小单拷贝区域(18,014 bp - 18,078 bp),由一对反向重复序列(25,377 - 25,427 bp)隔开。所有测序的质体基因组都有131个注释基因,包括86个蛋白质编码基因、8个rRNA基因和37个tRNA基因。将这五个新测序的叶绿体基因组与GenBank中保存的叶绿体基因组进行了比较。结果表明,所有叶绿体基因组在结构、内容和组织上高度相似。它们都呈现出四分体结构,并且所有基因和内含子的数量相同。密码子使用、RNA编辑位点和重复序列分析也显示这六个叶绿体基因组的结果高度相似。氨基酸亮氨酸的比例最高,几乎所有偏好的同义密码子都以A或U结尾。同样,在42个预测的RNA位点中,大多数转换是从丝氨酸(S)到亮氨酸(L)。检测到的大多数简单序列重复是A/T单核苷酸,使得叶绿体基因组富含A/T。IR边界的收缩和扩展也非常微小,因此对基因组大小差异的贡献很小。此外,序列变异分析表明,尽管鉴定出了几个潜在的条形码,如1、F和A,但叶绿体基因组具有几乎相同的基因组图谱。基于SNP和插入缺失分析,在 和 叶绿体序列中观察到更高的变异。系统发育重建进一步表明,这两个物种似乎是 的基部类群。紫茉莉属中很少栽培的野生种( )比常见栽培种和品种( 品种)分化得更早。总体而言,本研究的结果提供了额外的遗传资源,有助于紫茉莉属进一步的系统发育和进化研究。此外,本研究的遗传信息在鉴定紫茉莉属物种和品种方面可能有用,这对于分类学和植物育种研究都至关重要。