Center for Cancer Genomics and Precision Oncology, Wake Forest Baptist Comprehensive Cancer Center, Wake Forest Baptist Medical Center, Winston Salem, NC 27157, USA; Department of Cancer Biology, Wake Forest School of Medicine, Winston Salem, NC 27157, USA.
Center for Cancer Genomics and Precision Oncology, Wake Forest Baptist Comprehensive Cancer Center, Wake Forest Baptist Medical Center, Winston Salem, NC 27157, USA; Department of Biostatistics, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Genomics Proteomics Bioinformatics. 2021 Apr;19(2):330-341. doi: 10.1016/j.gpb.2020.09.002. Epub 2020 Dec 24.
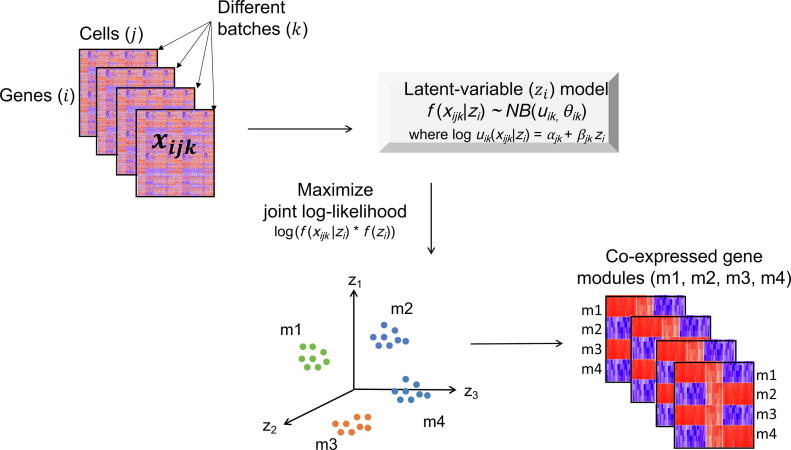
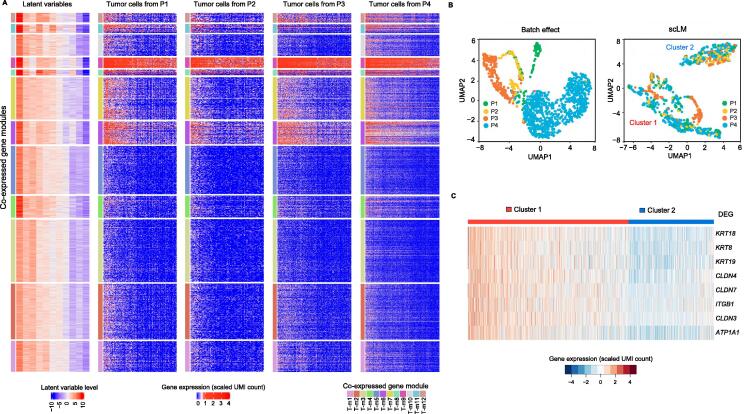
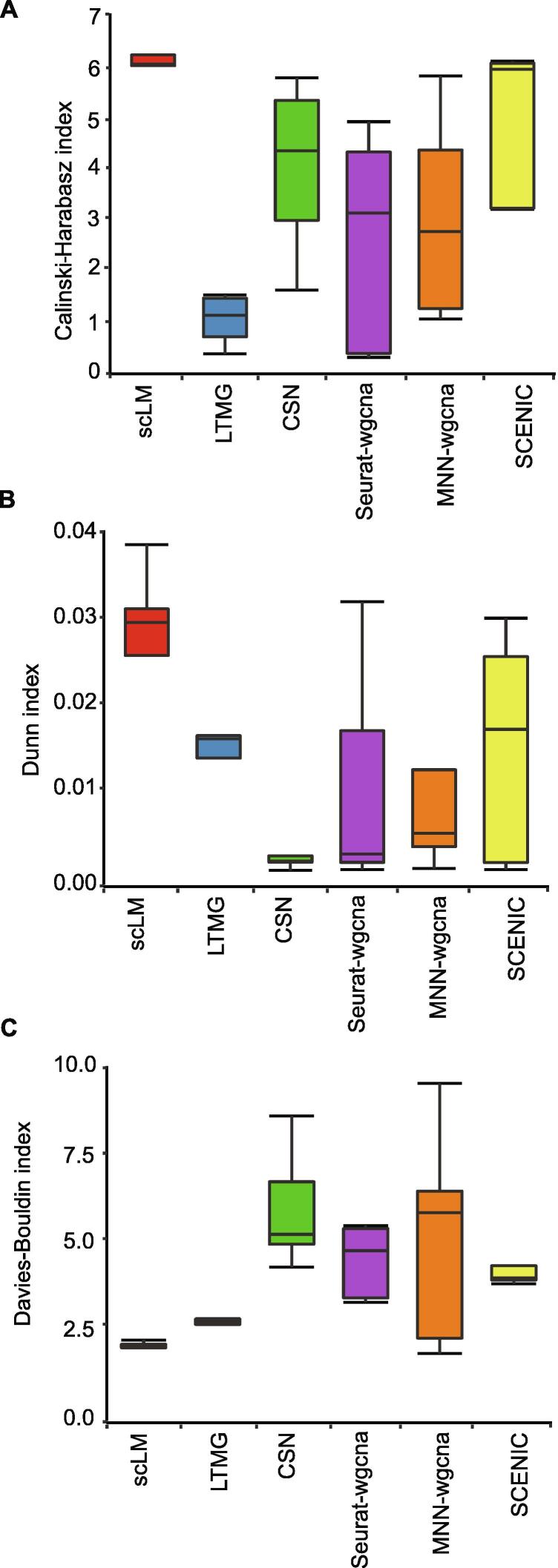
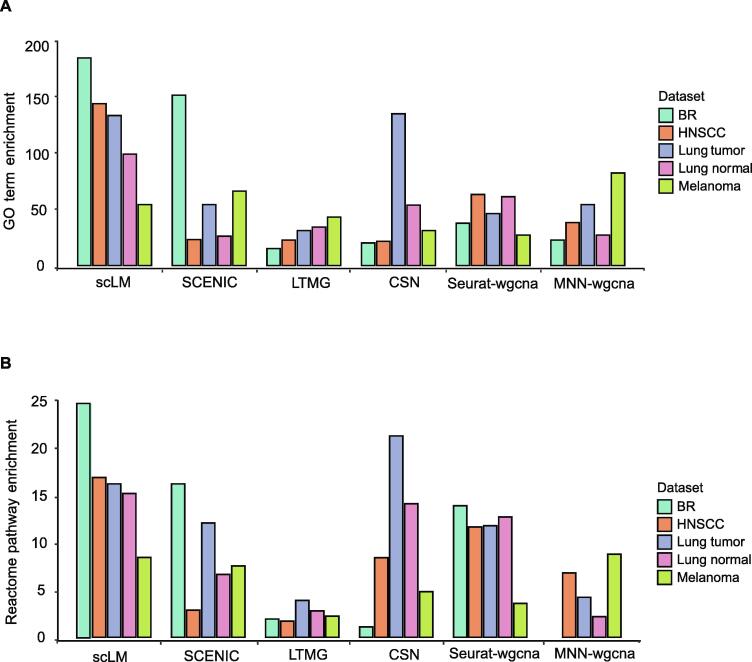
In gene expression profiling studies, including single-cell RNAsequencing (scRNA-seq) analyses, the identification and characterization of co-expressed genes provides critical information on cell identity and function. Gene co-expression clustering in scRNA-seq data presents certain challenges. We show that commonly used methods for single-cell data are not capable of identifying co-expressed genes accurately, and produce results that substantially limit biological expectations of co-expressed genes. Herein, we present single-cell Latent-variable Model (scLM), a gene co-clustering algorithm tailored to single-cell data that performs well at detecting gene clusters with significant biologic context. Importantly, scLM can simultaneously cluster multiple single-cell datasets, i.e., consensus clustering, enabling users to leverage single-cell data from multiple sources for novel comparative analysis. scLM takes raw count data as input and preserves biological variation without being influenced by batch effects from multiple datasets. Results from both simulation data and experimental data demonstrate that scLM outperforms the existing methods with considerably improved accuracy. To illustrate the biological insights of scLM, we apply it to our in-house and public experimental scRNA-seq datasets. scLM identifies novel functional gene modules and refines cell states, which facilitates mechanism discovery and understanding of complex biosystems such as cancers. A user-friendly R package with all the key features of the scLM method is available at https://github.com/QSong-github/scLM.
在基因表达谱研究中,包括单细胞 RNA 测序(scRNA-seq)分析,共同表达基因的鉴定和特征提供了关于细胞身份和功能的关键信息。单细胞 RNA-seq 数据中的基因共表达聚类存在一定的挑战。我们表明,常用的单细胞数据方法不能准确地识别共同表达的基因,并且产生的结果大大限制了对共同表达基因的生物学预期。在此,我们提出了单细胞潜在变量模型(scLM),这是一种专门针对单细胞数据的基因共聚类算法,在检测具有显著生物学背景的基因簇方面表现良好。重要的是,scLM 可以同时对多个单细胞数据集进行聚类,即共识聚类,使用户能够利用来自多个来源的单细胞数据进行新的比较分析。scLM 以原始计数数据为输入,保留了生物学变化,不受来自多个数据集的批次效应的影响。模拟数据和实验数据的结果表明,scLM 的准确性有了显著提高,优于现有方法。为了说明 scLM 的生物学见解,我们将其应用于我们内部和公共的实验单细胞 RNA-seq 数据集。scLM 确定了新的功能基因模块,并细化了细胞状态,这有助于发现机制并理解癌症等复杂生物系统。一个带有 scLM 方法所有关键特征的用户友好的 R 包可在 https://github.com/QSong-github/scLM 上获得。