Institut Curie, PSL Research University, Paris, France.
INSERM, U900, Paris, France.
PLoS Comput Biol. 2021 Jan 28;17(1):e1007900. doi: 10.1371/journal.pcbi.1007900. eCollection 2021 Jan.
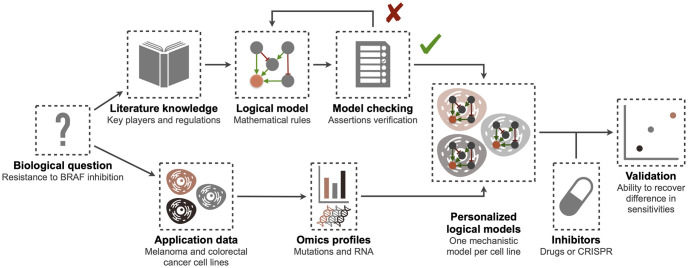
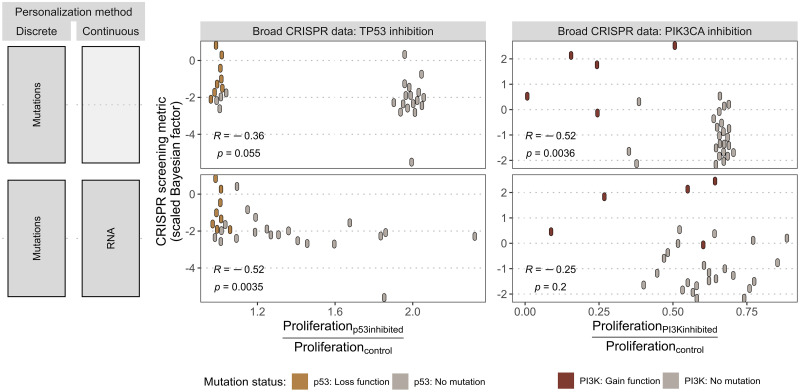
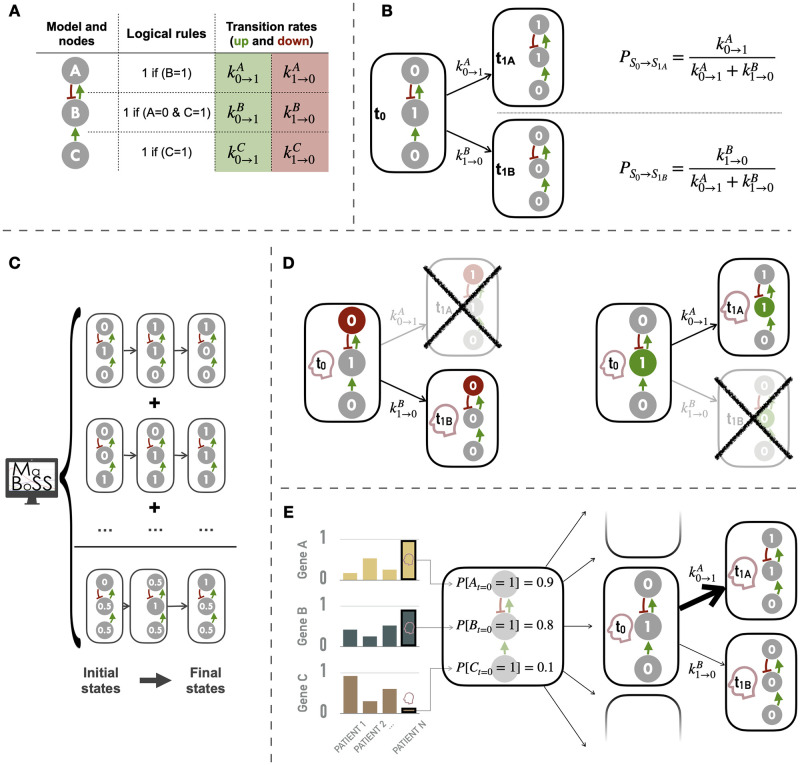
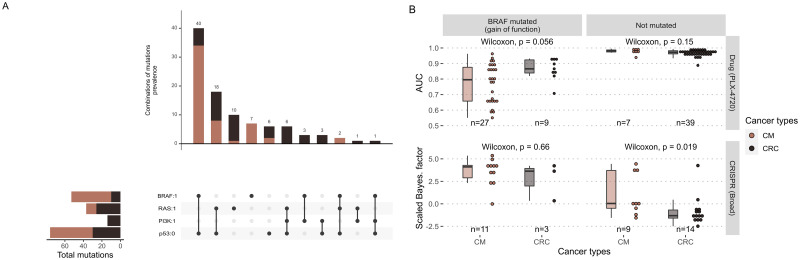
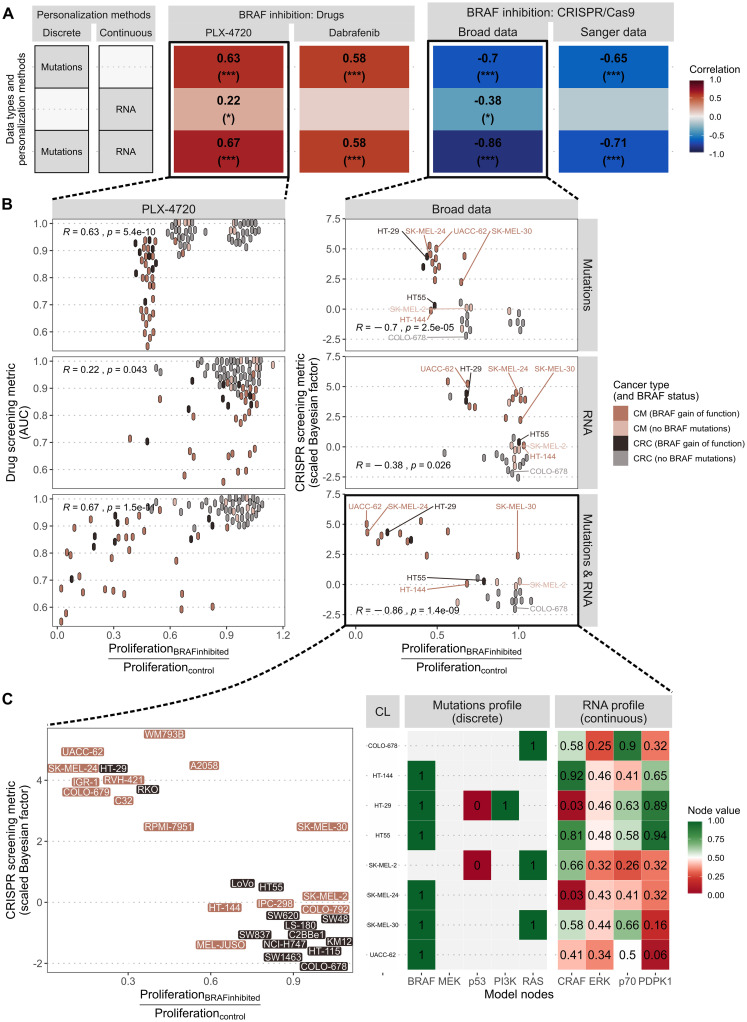
The study of response to cancer treatments has benefited greatly from the contribution of different omics data but their interpretation is sometimes difficult. Some mathematical models based on prior biological knowledge of signaling pathways facilitate this interpretation but often require fitting of their parameters using perturbation data. We propose a more qualitative mechanistic approach, based on logical formalism and on the sole mapping and interpretation of omics data, and able to recover differences in sensitivity to gene inhibition without model training. This approach is showcased by the study of BRAF inhibition in patients with melanomas and colorectal cancers who experience significant differences in sensitivity despite similar omics profiles. We first gather information from literature and build a logical model summarizing the regulatory network of the mitogen-activated protein kinase (MAPK) pathway surrounding BRAF, with factors involved in the BRAF inhibition resistance mechanisms. The relevance of this model is verified by automatically assessing that it qualitatively reproduces response or resistance behaviors identified in the literature. Data from over 100 melanoma and colorectal cancer cell lines are then used to validate the model's ability to explain differences in sensitivity. This generic model is transformed into personalized cell line-specific logical models by integrating the omics information of the cell lines as constraints of the model. The use of mutations alone allows personalized models to correlate significantly with experimental sensitivities to BRAF inhibition, both from drug and CRISPR targeting, and even better with the joint use of mutations and RNA, supporting multi-omics mechanistic models. A comparison of these untrained models with learning approaches highlights similarities in interpretation and complementarity depending on the size of the datasets. This parsimonious pipeline, which can easily be extended to other biological questions, makes it possible to explore the mechanistic causes of the response to treatment, on an individualized basis.
该研究通过不同的组学数据极大地促进了癌症治疗反应的研究,但对这些数据的解释有时很困难。一些基于信号通路先验生物学知识的数学模型有助于解释这些数据,但通常需要使用扰动数据来拟合它们的参数。我们提出了一种更定性的基于逻辑形式主义的机制方法,仅基于对组学数据的映射和解释,而无需模型训练,就能够恢复对基因抑制敏感性的差异。该方法通过对患有黑色素瘤和结直肠癌的患者中 BRAF 抑制作用的研究得到了展示,尽管这些患者的组学特征相似,但他们对 BRAF 抑制的敏感性存在显著差异。我们首先从文献中收集信息,并构建一个逻辑模型,总结 BRAF 周围的丝裂原活化蛋白激酶 (MAPK) 通路的调控网络,其中包括涉及 BRAF 抑制耐药机制的因素。通过自动评估该模型定性再现文献中识别出的反应或耐药行为,验证了该模型的相关性。然后使用来自 100 多个黑色素瘤和结直肠癌细胞系的数据来验证该模型解释敏感性差异的能力。通过将细胞系的组学信息作为模型的约束条件,将通用模型转化为个性化的细胞系特定逻辑模型。仅使用突变就可以使个性化模型与 BRAF 抑制的实验敏感性(包括药物和 CRISPR 靶向)显著相关,甚至与突变和 RNA 的联合使用相关性更好,支持多组学机制模型。与学习方法相比,这些未经训练的模型的比较突出了根据数据集大小进行解释的相似性和互补性。这种简洁的方法可以很容易地扩展到其他生物学问题,使我们能够在个体化的基础上探索对治疗反应的机制原因。