Pathogen Discovery Laboratory, Institut Pasteur, 75015 Paris, France.
Bioinformatics and Biostatistics Hub, Computational Biology Department, Institut Pasteur, 75015 Paris, France.
Viruses. 2021 Feb 7;13(2):253. doi: 10.3390/v13020253.
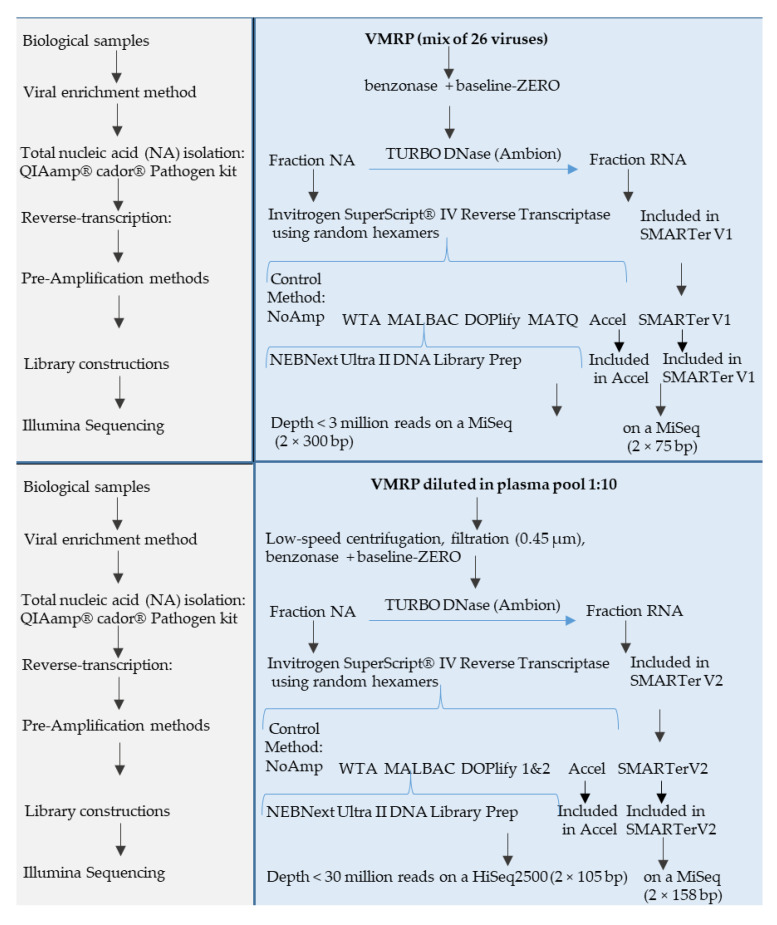
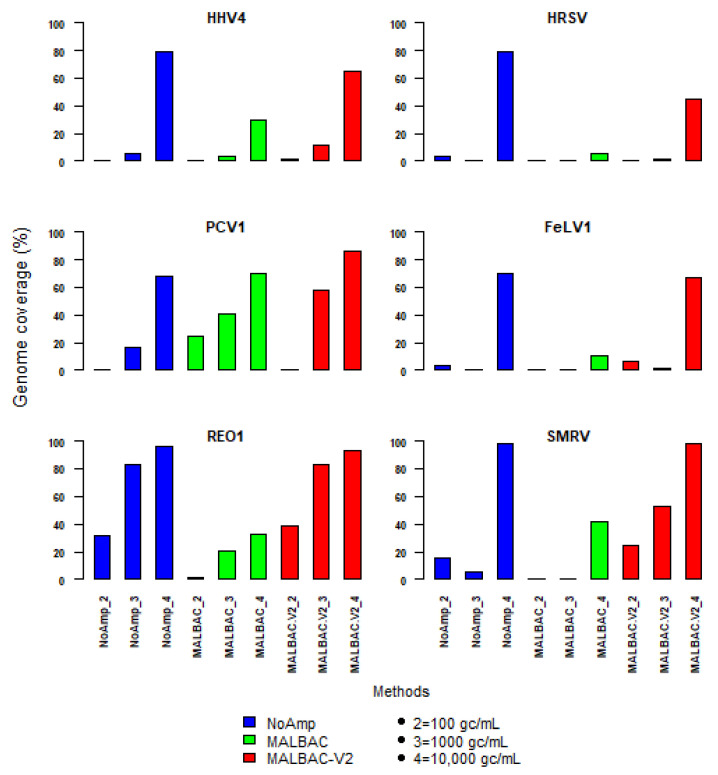
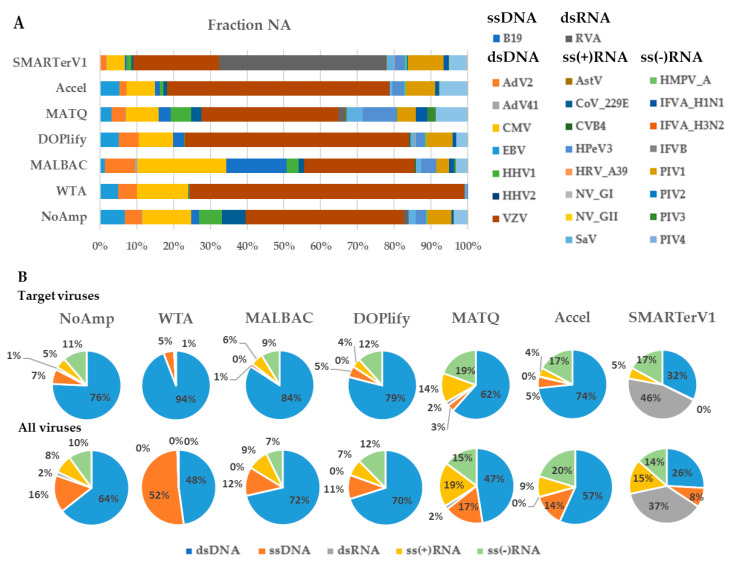
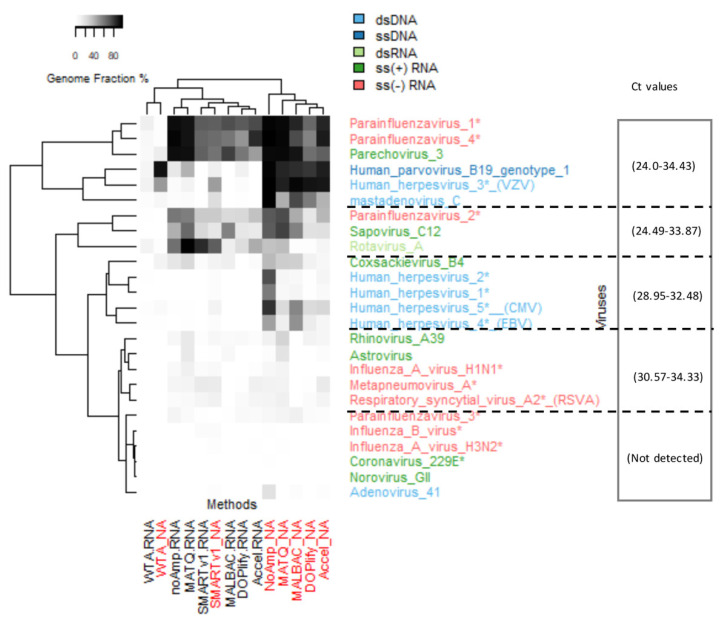
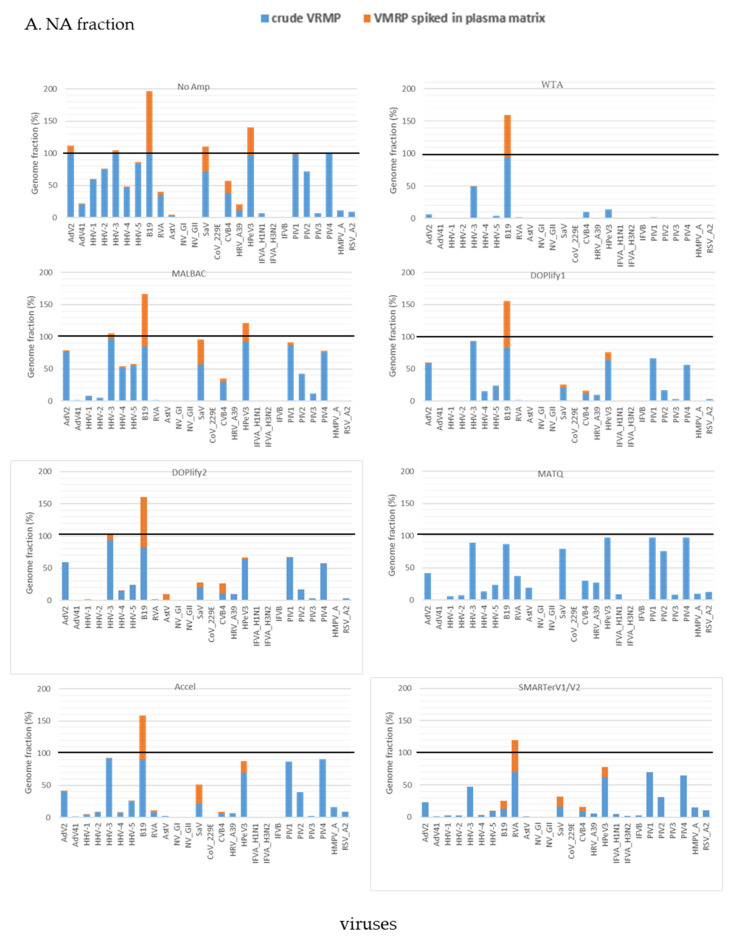
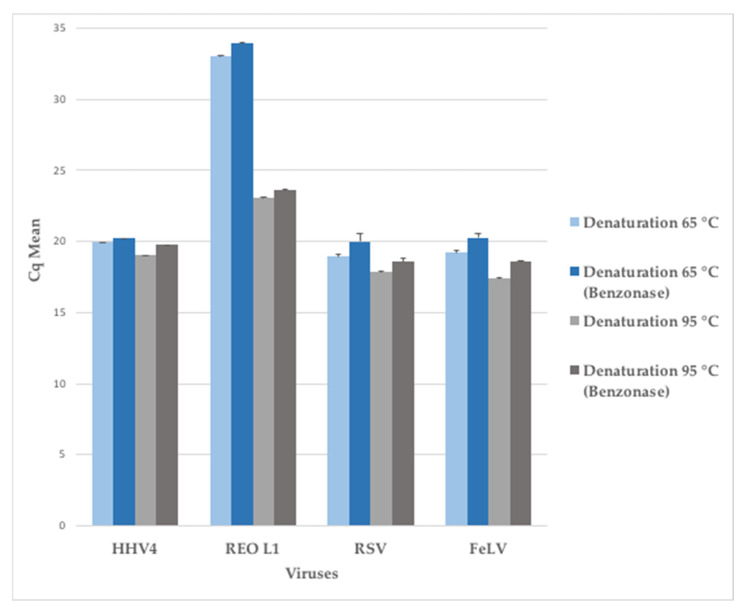
Clinical metagenomics is a broad-range agnostic detection method of pathogens, including novel microorganisms. A major limit is the low pathogen load compared to the high background of host nucleic acids. To overcome this issue, several solutions exist, such as applying a very high depth of sequencing, or performing a relative enrichment of viral genomes associated with capsids. At the end, the quantity of total nucleic acids is often below the concentrations recommended by the manufacturers of library kits, which necessitates to random amplify nucleic acids. Using a pool of 26 viruses representative of viral diversity, we observed a deep impact of the nature of sample (total nucleic acids versus RNA only), the reverse transcription, the random amplification and library construction method on virus recovery. We further optimized the two most promising methods and assessed their performance with fully characterized reference virus stocks. Good genome coverage and limit of detection lower than 100 or 1000 genome copies per mL of plasma, depending on the genome viral type, were obtained from a three million reads dataset. Our study reveals that optimized random amplification is a technique of choice when insufficient amounts of nucleic acid are available for direct libraries constructions.
临床宏基因组学是一种广泛的病原体无偏检测方法,包括新型微生物。主要限制是与宿主核酸的高背景相比,病原体载量较低。为了克服这个问题,有几种解决方案,例如应用非常高的测序深度,或对与衣壳相关的病毒基因组进行相对富集。最后,总核酸的数量通常低于文库试剂盒制造商推荐的浓度,这需要随机扩增核酸。使用代表病毒多样性的 26 种病毒池,我们观察到样本性质(总核酸与仅 RNA)、逆转录、随机扩增和文库构建方法对病毒回收的深刻影响。我们进一步优化了两种最有前途的方法,并使用完全特征化的参考病毒株评估了它们的性能。从三百万个读数数据集获得了良好的基因组覆盖率和检测限,低于 100 或 1000 个基因组拷贝/毫升血浆,具体取决于病毒类型。我们的研究表明,当可用于直接文库构建的核酸量不足时,优化的随机扩增是一种首选技术。