Institute of Molecular, Cell and Systems Biology, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, UK.
Glasgow Polyomics, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, UK.
J Huntingtons Dis. 2021;10(1):53-74. doi: 10.3233/JHD-200433.
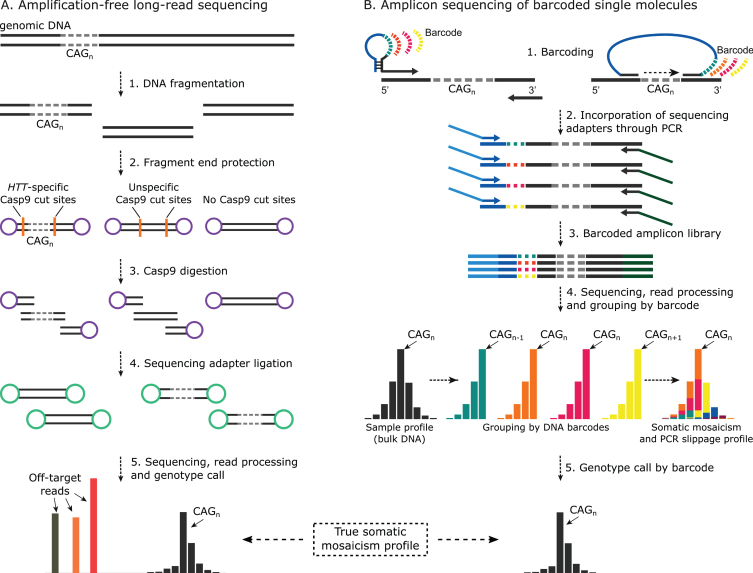
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by the expansion of the HTT CAG repeat. Affected individuals inherit ≥36 repeats and longer alleles cause earlier onset, greater disease severity and faster disease progression. The HTT CAG repeat is genetically unstable in the soma in a process that preferentially generates somatic expansions, the proportion of which is associated with disease onset, severity and progression. Somatic mosaicism of the HTT CAG repeat has traditionally been assessed by semi-quantitative PCR-electrophoresis approaches that have limitations (e.g., no information about sequence variants). Genotyping-by-sequencing could allow for some of these limitations to be overcome.
To investigate the utility of PCR sequencing to genotype large (>50 CAGs) HD alleles and to quantify the associated somatic mosaicism.
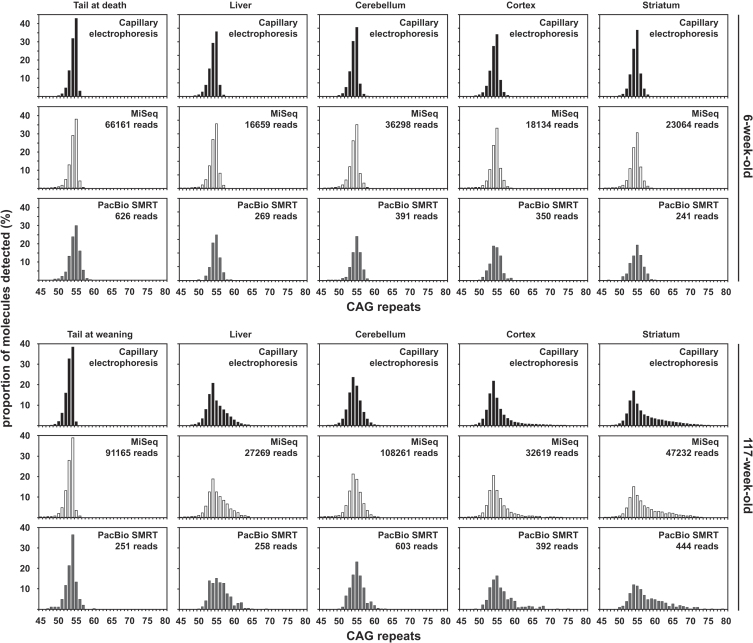
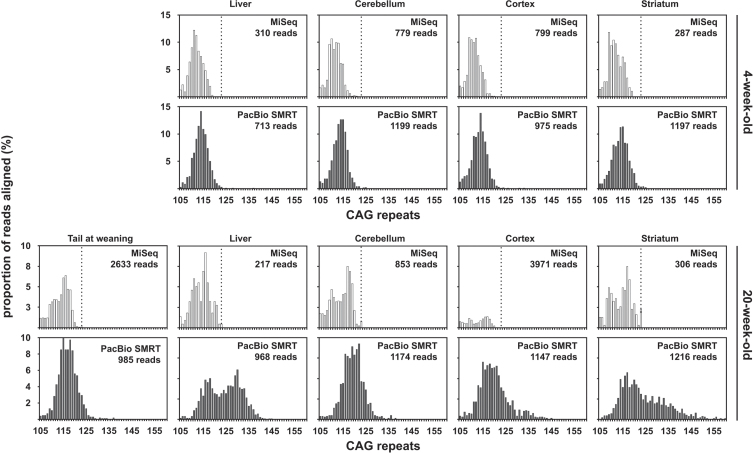
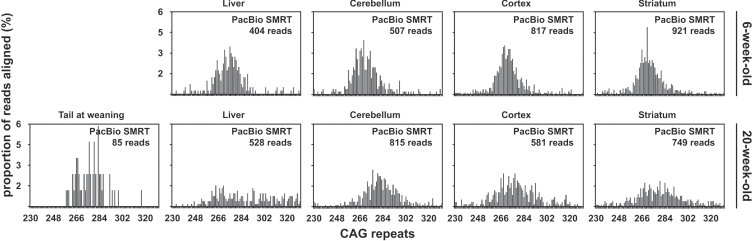
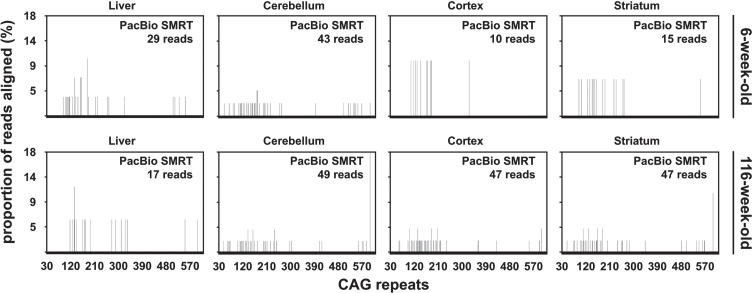
We have applied MiSeq and PacBio sequencing to PCR products of the HTT CAG repeat in transgenic R6/2 mice carrying ∼55, ∼110, ∼255 and ∼470 CAGs. For each of these alleles, we compared the repeat length distributions generated for different tissues at two ages.
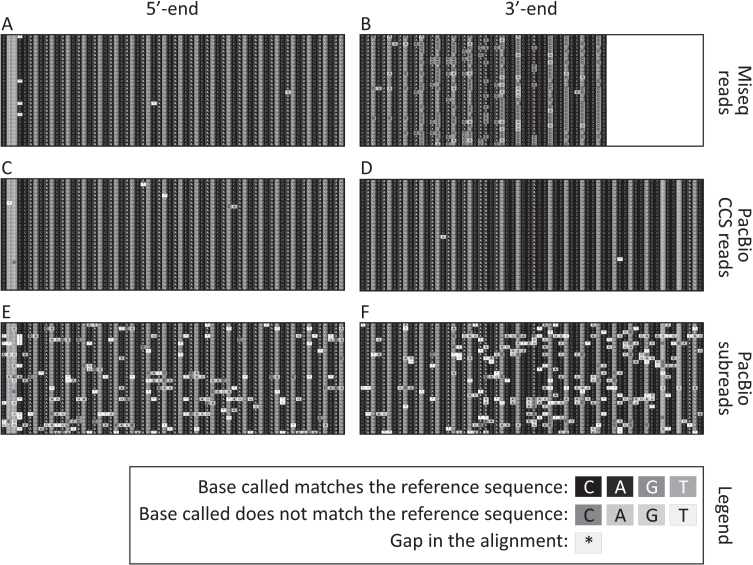
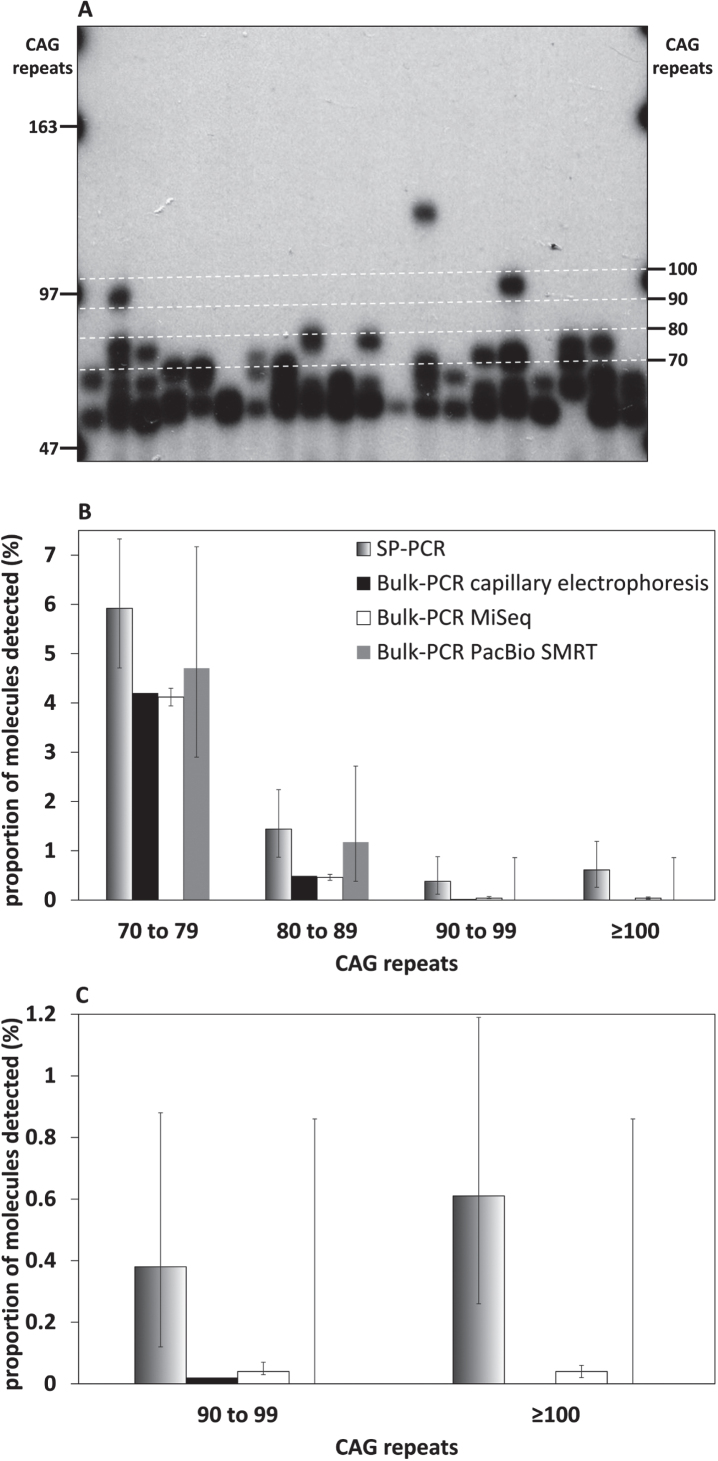
We were able to sequence the CAG repeat full length in all samples. However, the repeat length distributions for samples with ∼470 CAGs were biased towards shorter repeat lengths.
PCR sequencing can be used to sequence all the HD alleles considered, but this approach cannot be used to estimate modal allele size or quantify somatic expansions for alleles ⪢250 CAGs. We review the limitations of PCR sequencing and alternative approaches that may allow the quantification of somatic contractions and very large somatic expansions.
亨廷顿病(HD)是一种常染色体显性神经退行性疾病,由 HTT CAG 重复扩展引起。受影响的个体遗传了≥36 个重复,更长的等位基因导致发病更早、疾病严重程度更高、疾病进展更快。在体细胞中,HTT CAG 重复在遗传上不稳定,这一过程优先产生体细胞扩展,其比例与疾病发病、严重程度和进展相关。HTT CAG 重复的体细胞镶嵌传统上通过半定量 PCR-电泳方法进行评估,这些方法存在局限性(例如,没有关于序列变异的信息)。基于测序的基因分型可能可以克服其中一些局限性。
研究 PCR 测序在对大型(>50 CAGs)HD 等位基因进行基因分型和量化相关体细胞镶嵌方面的应用。
我们已经将 MiSeq 和 PacBio 测序应用于携带约 55、110、255 和 470 CAG 的 R6/2 转基因小鼠的 HTT CAG 重复 PCR 产物。对于每个这些等位基因,我们比较了两个年龄的不同组织产生的重复长度分布。
我们能够对所有样本的 CAG 重复全长进行测序。然而,具有约 470 CAG 的样本的重复长度分布偏向于较短的重复长度。
PCR 测序可用于对所有考虑的 HD 等位基因进行测序,但这种方法不能用于估计模态等位基因大小或量化等位基因>250 CAG 的体细胞扩展。我们回顾了 PCR 测序的局限性以及可能允许量化体细胞收缩和非常大的体细胞扩展的替代方法。