Ludwig Institute for Cancer Research, Nuffield Department of Medicine, University of Oxford, Oxford OX3 7FZ, UK.
Target Discovery Institute, Nuffield Department of Medicine, University of Oxford, Oxford OX3 7FZ, UK.
Nucleic Acids Res. 2021 Jul 21;49(13):e76. doi: 10.1093/nar/gkab291.
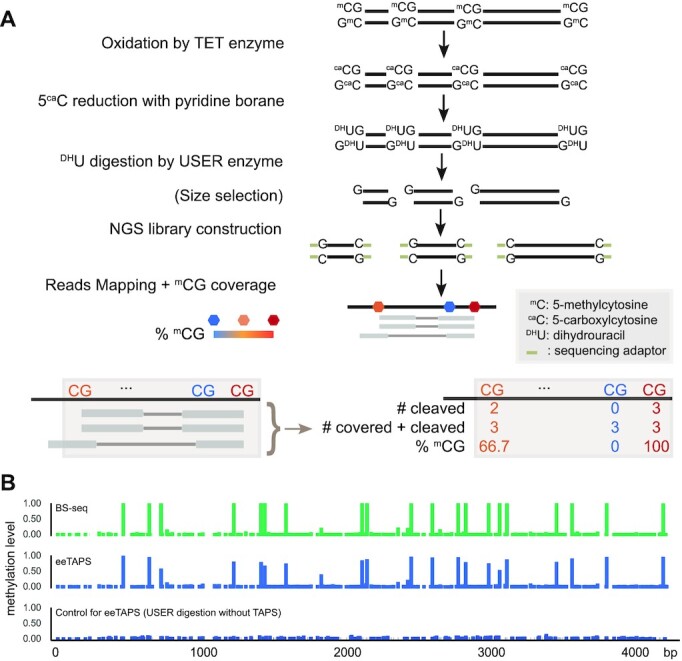
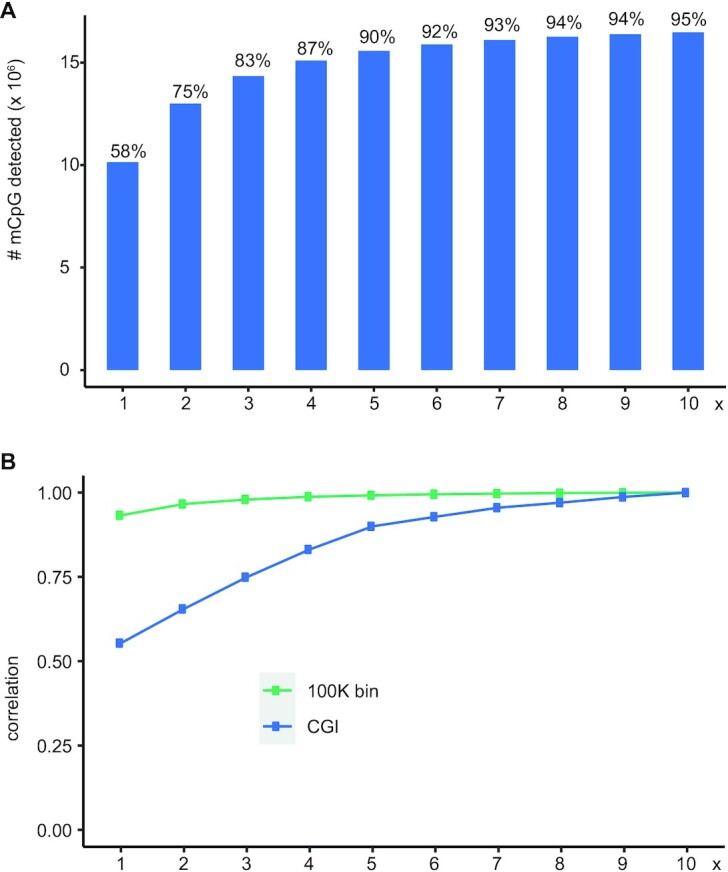
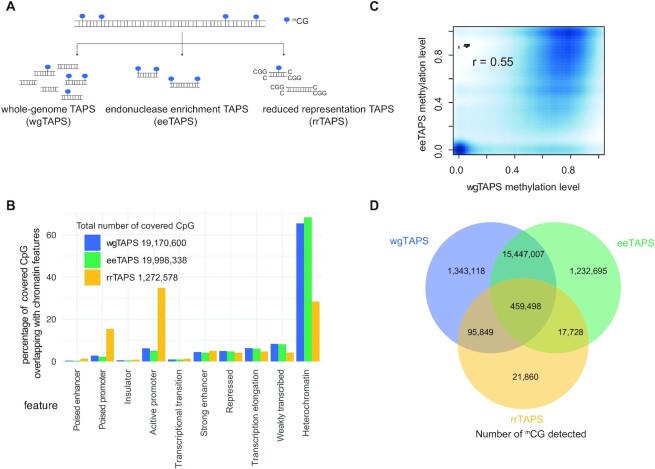
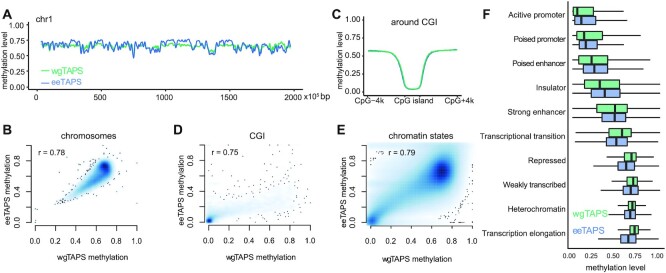
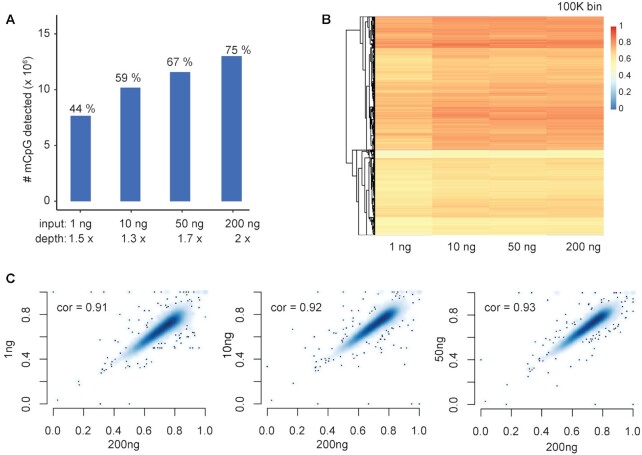
Whole genome base-resolution methylome sequencing allows for the most comprehensive analysis of DNA methylation, however, the considerable sequencing cost often limits its applications. While reduced representation sequencing can be an affordable alternative, over 80% of CpGs in the genome are not covered. Building on our recently developed TET-assisted pyridine borane sequencing (TAPS) method, we here described endonuclease enrichment TAPS (eeTAPS), which utilizes dihydrouracil (DHU)-cleaving endonuclease digestion of TAPS-converted DNA to enrich methylated CpG sites (mCpGs). eeTAPS can accurately detect 87% of mCpGs in the mouse genome with a sequencing depth equivalent to 4× whole genome sequencing. In comparison, reduced representation TAPS (rrTAPS) detected less than 4% of mCpGs with 2.5× sequencing depth. Our results demonstrate eeTAPS to be a new strategy for cost-effective genome-wide methylation analysis at single-CpG resolution that can fill the gap between whole-genome and reduced representation sequencing.
全基因组碱基分辨率甲基化测序可以对 DNA 甲基化进行最全面的分析,然而,相当大的测序成本通常限制了其应用。虽然降低代表性测序是一种经济实惠的替代方法,但基因组中超过 80%的 CpG 未被覆盖。基于我们最近开发的 TET 辅助吡啶硼烷测序(TAPS)方法,我们在这里描述了内切酶富集 TAPS(eeTAPS),它利用 TAPS 转化的 DNA 中二氢尿嘧啶(DHU)切割内切酶消化来富集甲基化 CpG 位点(mCpGs)。eeTAPS 可以准确地检测到小鼠基因组中 87%的 mCpGs,其测序深度相当于 4 倍全基因组测序。相比之下,降低代表性 TAPS(rrTAPS)在 2.5 倍测序深度下检测到的 mCpGs 不到 4%。我们的结果表明,eeTAPS 是一种在单 CpG 分辨率下进行经济高效的全基因组甲基化分析的新策略,可以填补全基因组和降低代表性测序之间的空白。