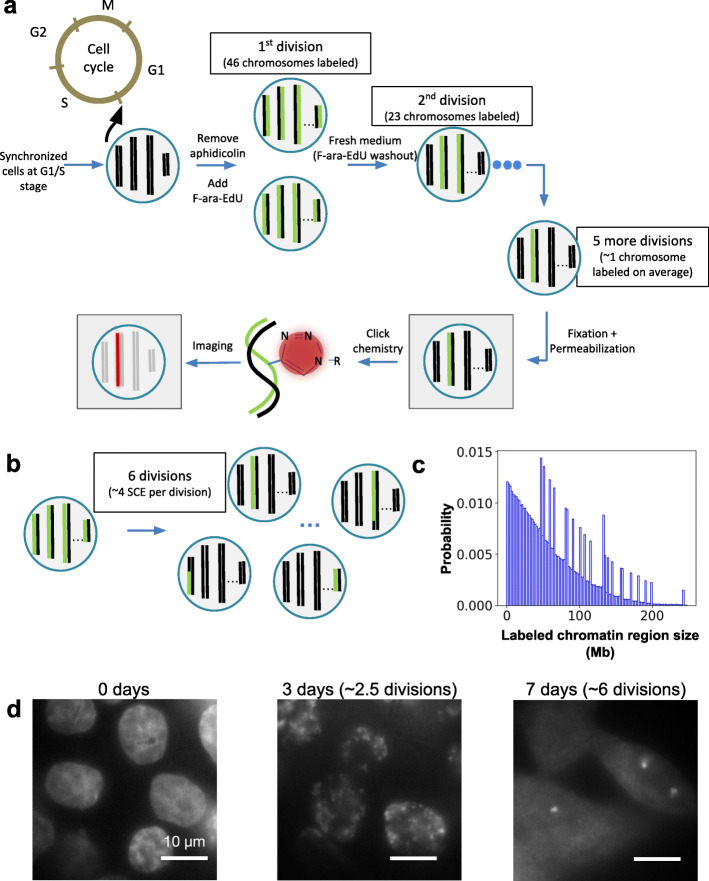
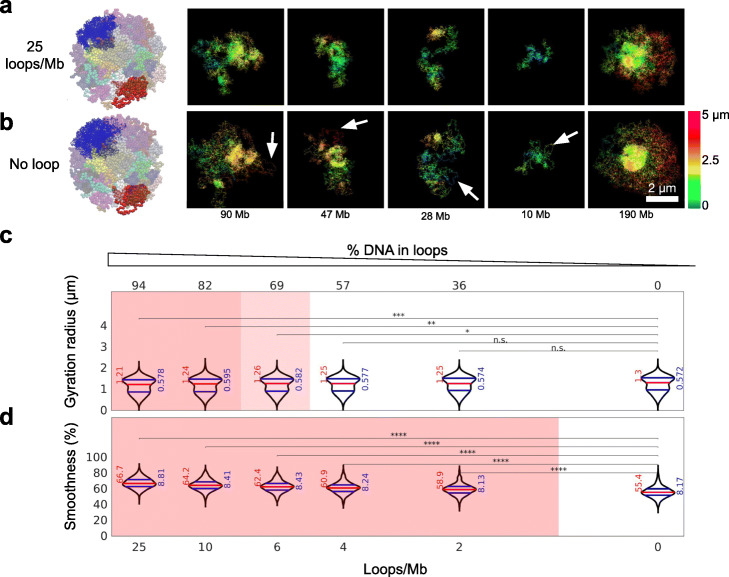
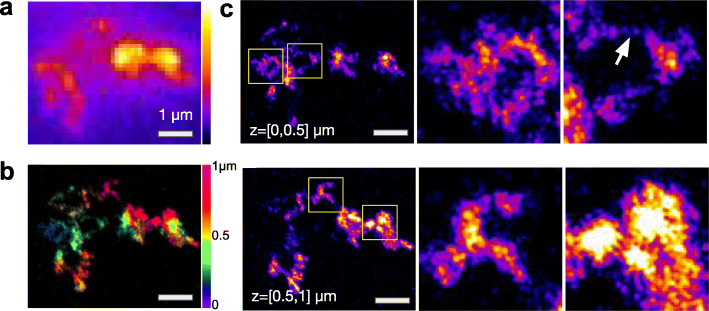
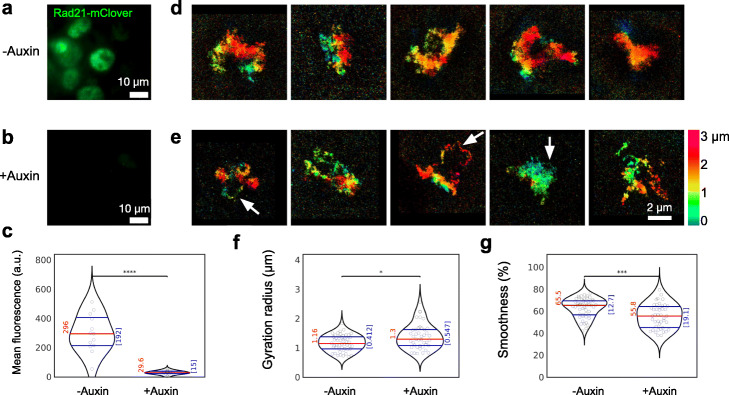
Institut Pasteur, Imaging and Modeling Unit, UMR 3691, CNRS, Paris, France.
School of Public Health & Jiangxi Provincial Key Laboratory of Preventive Medicine, Nanchang University, Nanchang, 330006, China.
Genome Biol. 2021 May 11;22(1):150. doi: 10.1186/s13059-021-02343-w.
The 3D organization of the chromatin fiber in cell nuclei plays a key role in the regulation of gene expression. Genome-wide techniques to score DNA-DNA contacts, such as Hi-C, reveal the partitioning of chromosomes into epigenetically defined active and repressed compartments and smaller "topologically associated" domains. These domains are often associated with chromatin loops, which largely disappear upon removal of cohesin. Because most Hi-C implementations average contact frequencies over millions of cells and do not provide direct spatial information, it remains unclear whether and how frequently chromatin domains and loops exist in single cells.
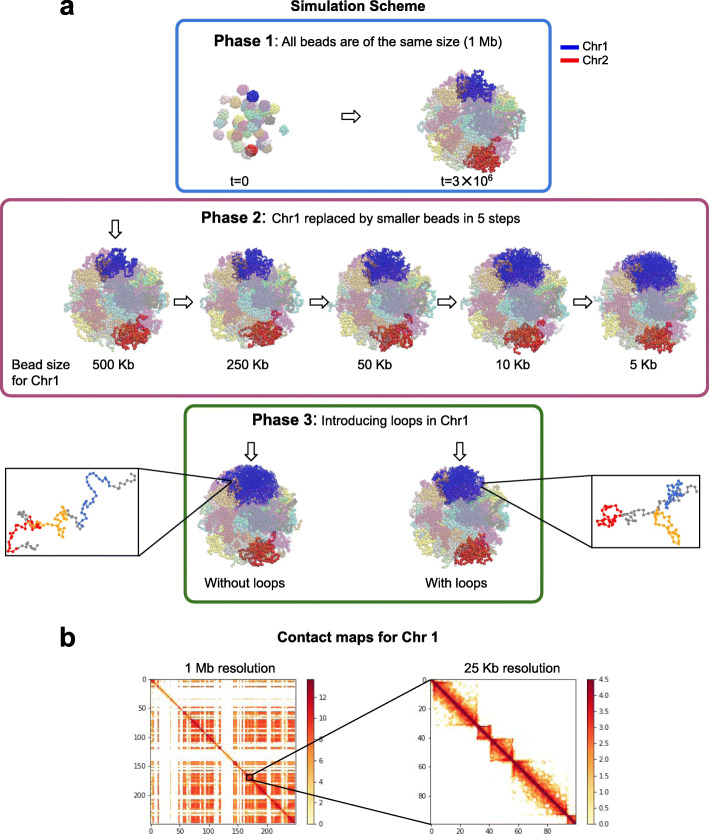
We combine 3D single-molecule localization microscopy with a low-cost fluorescence labeling strategy that does not denature the DNA, to visualize large portions of single human chromosomes in situ at high resolution. In parallel, we develop multi-scale, whole nucleus polymer simulations, that predict chromatin structures at scales ranging from 5 kb up to entire chromosomes. We image chromosomes in G1 and M phase and examine the effect of cohesin on interphase chromatin structure. Depletion of cohesin leads to increased prevalence of loose chromatin stretches, increased gyration radii, and reduced smoothness of imaged chromatin regions. By comparison to model predictions, we estimate that 6-25 or more purely cohesin-dependent chromatin loops coexist per megabase of DNA in single cells, suggesting that the vast majority of the genome is enclosed in loops.
Our results provide new constraints on chromatin structure and showcase an affordable non-invasive approach to study genome organization in single cells.
细胞核中染色质纤维的 3D 组织在基因表达调控中起着关键作用。用于评估 DNA-DNA 接触的全基因组技术,如 Hi-C,揭示了染色体在表观遗传定义的活跃和抑制隔室以及较小的“拓扑相关”域中的分区。这些域通常与染色质环相关联,而这些环在去除凝聚蛋白后大部分消失。由于大多数 Hi-C 实现平均接触频率在数百万个细胞上,并且不提供直接的空间信息,因此仍然不清楚染色质域和环是否以及在何种程度上存在于单个细胞中。
我们结合了 3D 单分子定位显微镜和一种低成本的荧光标记策略,该策略不会使 DNA 变性,从而以高分辨率原位可视化大片段的单个人类染色体。同时,我们开发了多尺度的整个核聚合物模拟,可预测从 5kb 到整个染色体的范围内的染色质结构。我们在 G1 和 M 期对染色体进行成像,并研究了凝聚蛋白对间期染色质结构的影响。凝聚蛋白的耗竭导致松散染色质伸展的出现频率增加、回旋半径增加以及成像染色质区域的平滑度降低。与模型预测相比,我们估计在单个细胞中每兆碱基 DNA 中存在 6-25 个或更多纯依赖于凝聚蛋白的染色质环,这表明绝大多数基因组都被包含在环中。
我们的结果为染色质结构提供了新的限制,并展示了一种经济实惠的非侵入性方法来研究单个细胞中的基因组组织。